Introduction
Currently, one of the most substantial threats to the terrestrial ecosystems is the rapid decline in biodiversity, which has become a particularly important focus of nature conservation initiatives. In this regard, invasive species are considered among the most prominent causes of biodiversity loss, second only to habitat destruction or transformation. Invaded ecosystems are typically damaged to varying degrees by non-native species via changes in species diversity and community structures. Non-native species that spread rapidly over extensive areas, and thereby threaten local biological diversity, are considered invasive species (Genovesi & Shine,2004). In worst case scenarios, the mass distribution of new organisms can lead to local extinctions among native species (Vilà et al.,2007). By cultivating alien plants in residential and botanical gardens, humans unwittingly contribute to the spread of these plants. Cultivated taxa capable of self-propagation can readily disperse into natural environments, some of which are able to establish populations in different habitats. Breeding or hybridization in gardens can even alter the genetic make-up of species, and consequently, escaped specimens might have already undergone adaptive changes. Adequate knowledge of the origin and distribution of alien plant species and the ecology of populations is crucial for the conservation of biodiversity (Bevill & Louda,1999; Duncan & Young,2000).
The genus Hedera is the single representative of the Araliaceae plant family in Europe. According to McAllister and Marshall (2017) and Valcárcel et al. (2017), the genus comprises 12 evergreen-vine species distributed across the Northern Hemisphere, from Macaronesia, Europe, and North Africa to South and East Asia. It is well known that Hedera species are difficult to identify, owing to the diverse morphological characters of their leaves, which can be affected by environmental conditions and differ according to ontogenic stage during individual development. Indeed, there can be significant differences in the appearance between the juvenile and adult shoots of an individual, and ontogenic changes are known to be reversible (Robbins,1957,1960; Rogler & Hackett,1975).
The first attempts to classify Hedera were based on morphological traits of the vegetative organs, for which leaf morphology and the foliar trichomes were used as the main diagnostic characters in species delimitation. Subsequently, however, trichome morphology was shown to be not decisive trait for the delimitation of different taxa, as these features were considered evolutionary unstable (Green et al.,2011; Vargas et al.,1999). During the early 1980s, cytological analyses were introduced in taxonomical studies. For example, the separation of H. hibernica from H. helix, which were previously assumed to be taxonomically identical, was based on the different levels of ploidy (H. hibernica is a tetraploid, whereas H. helix is a diploid) in addition to differences in trichomes (Jacobsen,1954; McAllister & Rutherford,1990). Hedera algeriensis (2x) and H. maroccana (4x) were similarly differentiated (Rutherford,1984,1989). Nonetheless, based on area disjunction that has promoted geographic isolation, H. iberica and H. maderensis (Ackerfield & Wen,2003), and H. nepalensis var. nepalensis and H. nepalensis var. sinensis have been distinguished (Ackerfield,2001; Ackerfield & Wen,2002). Polyploidy is a frequent phenomenon in the genus Hedera, in which diploid, tetraploid, hexaploid, and octoploid populations are known (Vargas et al.,1999), and polyploidization events followed by geographic isolation have been identified as prominent processes in promoting the diversification of species (Valcárcel et al.,2017).
On the basis of trichome morphology, geographical distribution, and ploidy level, four species of Hedera have been recognized in Asia, namely, H. colchica Koch (8x), H. nepalensis Koch (2x), H. pastuchovii Woronow (6x), and H. rhombea (Miq.) Bean (2x), all of which are characterized by scale-like trichomes. Furthermore, in Central Europe, two species with stellate trichomes are known, namely, H. helix L. (2x), which has multiangulate erect trichomes, and H. hibernica (Kirch.) Bean (4x), which bears bifurcate stellate trichomes. The most diverse region in terms of Hedera species is the Mediterranean region, in which the following seven taxa have been described: H. azorica Carr. (2x), which has stellate hairs, and H. algeriensis Hibberd (4x), H. canariensis Willd (2x), H. iberica McAllister (6x), H. maderensis Koch ex Rutherford (6x), H. maroccana (2x), and H. pastuchovii ssp. cypria McAllister (6x), which all possess scale-like trichomes (McAllister & Marshall,2017).
In addition to numerous morphological studies (Ackerfield,2001; Ackerfied & Wen,2002,2003; Kost et al.,2003; Lum & Maze,1989; Valcárcel & Vargas,2010), molecular studies, which started in the late 1990s, have provided new insights into taxonomical relationships, phylogeography, and evolution of the genus Hedera (Ackerfield & Wen,2003; Green et al.,2011; Grivet & Petit,2002; Lowry et al.,2004; Valcárcel et al.,2003,2017; Valcárcel & Vargas,2012; Vargas et al.,1999). Phylogenetic and phylogeographic studies have confirmed Asia to be the center of origin for the genus, with the Mediterranean area being considered a secondary center of diversification (Valcárcel et al.,2003,2017; Valcárcel & Vargas,2012). However, these molecular studies have not been able to conclusively clarify the phylogenetic lineages of all taxa, owing to the low genetic variability between species within the genus.
In Central Europe, H. helix is considered to be the only native ivy (Rose,1996; Soó & Jávorka,1951). In addition to H. helix, however, several species are mass cultivated and have horticultural importance, including the Western European H. hibernica and the Central Asian H. colchica. Furthermore, H. azorica, H. maroccana, and H. rhombea are cultivated to a lesser extent, although mainly in private collections or botanical gardens. However, during the early 2000s, the spread of an ivy that differs from the known species with respect to several traits was detected in Hungary (Bényei-Himmer et al.,2017). Initially, this taxon was regarded to be a variety of the tetraploid H. hibernica (Udvardy & Bényei-Himmer,1999), although subsequent cytological analyses (Lengyel et al.,2007) revealed it to be diploid (2x = 48). On the basis of an assessment of morphological, cytological, and phenological traits, the newly identified species has been described under the name H. crebrescens (Bényei-Himmer et al.,2017). According to field observations, this species is spreading spontaneously in several parts of Hungary, and has also been observed in other neighboring countries, namely, Western Ukraine, Slovakia, Austria, Germany, and The Netherlands. Owing to its high reproductive capacity (high rate of viable seed production, successful propagation by birds, and high germination rate) and vigorous growth, this species is considered to be a potentially invasive plant that is becoming naturalized in many parts of Central Europe, thereby threatening the habitats of the autochthonous H. helix (Bényei-Himmer et al.,2017). Indeed, this newly identified species has recently been included in the black list of the Hungarian flora (Bartha,2020, p. 20).
With reference to earlier phylogenetic studies on the genus, we conducted a molecular study in which we sought to reveal the taxonomical position and phylogenetic relationships of Hedera crebrescens. Highlighting the importance of species’ identification, we aimed at distinguishing this species from other ivy taxa to prevent its excessive growth and invasive spread throughout the habitats of the native H. helix.
Material and Methods
Plant Material and GenBank Sequences
Hedera crebrescens Himmer-Höhn, is a diploid taxon that was first described in 2017 by Bényei-Himmer and coauthors (Bényei-Himmer et al.,2017), after a 20-year-long survey of specimens. Hedera crebrescens can be distinguished from its presumed closest relatives H. helix and varieties of H. hibernica based on certain pronounced vegetative and generative traits. Hedera crebrescens has characteristic reddish green shoots. Leaves on the horizontally creeping shoots are triangular, slightly trilobed with a cordate leaf base, whereas those on the vertically emerging shoots are large-lobed (three to five; 10–16 cm) with overlapping bases (Figure 1). On the vegetative shoots, the apical lobes of the leaves are wide and dome-shaped, whereas leaves on the generative shoots are heart-shaped with a cordate leaf base. The stellate trichomes are multiangulate (7–12 rays) and typically appressed. The inflorescence is short (3–5 cm) with only a few or no lateral umbels. In the apical umbels, the flowers are crowded. Beneath the apical umbel, there is a solitary flower that rarely produces fruit. The densely packed fruits are black and contain three–five seeds. Hedera crebrescens also differs from H. helix and H. hibernica with respect to its vigorous growth and the slight frost sensitivity of its leaves (Bényei-Himmer et al.,2017).

From the total of 63 Hedera samples that were examined in the present study, we sampled eight specimens: five samples of H. crebrescens and one of each of the three assumed relatives H. helix, H. hibernica, and H. azorica. The samples were selected from the Ivy Collections of the Soroksár Botanical Garden, the Buda Arboretum in Budapest, the downtown area of Budapest, and the outskirts of the city of Zebegény, Hungary (also see Table S1). Sequences of these samples were aligned with Hedera sequences from the GenBank reported in previous studies (Table S2). For plastid regions, we selected 26 sequences from Green et al. (2011), whereas for nuclear regions, we selected 27 sequences from Vargas et al. (1999), three sequences from Valcárcel et al. (2003), and one sequences from Li et al. (2013). In this regard, it should be mentioned that the samples used for the chloroplast and nuclear analyses in these previous studies are not identical.
DNA Extraction, Amplification, and Sequencing
Total genomic DNA was extracted from fresh leaves (20–25 mg) using a DNeasy Mini Kit (QIAGEN, Valencia, CA, USA) following the manufacturer’s recommendations. With reference to the phylogenetic study conducted by Green et al. (2011), we used universal plastid primers to amplify five noncoding chloroplast regions, namely, the matk (psbA5′R-matk8F) intergenic spacer, trnH-psbA (psbA-trnHGUG) intergenic spacer, psbB-psbH (psbB-psbH) intergenic spacer, rpl16 (rpL16F71-rpL16R1516) intron, and rpoB-trnCGCA (rpoB-trnCGCAR) intergenic spacer (Shaw et al.,2005; Taberlet et al.,1991). With reference to Vargas et al. (1999), we amplified the internal transcribed spacer ITS1-5.8S-ITS2 region using the ITS5/ITS4 primer pair (White et al.,1990) (Table 1).
Table 1
Description and amplification conditions of the six primer pairs (five chloroplast and one nuclear) used for the molecular study.
PCR amplifications were performed using 24.5-μL reactions mixture containing 1 μL genomic DNA (20–30 ng), 18.7 μL Milli-Q ultrapure water (Merckmillipore, Billerica, MA, USA), 2.5 μL (10×) Dream Taq Green PCR buffer (ThermoFisher, Waltham, MA, USA), 0.5 μL (10 mM) dNTP mix (ThermoFisher), 1 μL (2.5 mM) MgCl2, 0.5 μL (10 mM) of each primer (Biocenter Kft., Szeged, Hungary), 0.25 μL (1%) bovine serum albumin (ThermoFisher), 0.5 μL (2%) dimethyl sulfoxide (Reanal, Budapest, Hungary), and 0.05 μL (0.5 unit) Dream Taq GREEN DNA polymerase (ThermoFisher).
The thermocycling conditions used for amplification of the plastid regions were based on those described by Ramsey et al. (2005) with some modifications. In the case of certain primers, it was necessary to optimize the protocols for gradient PCR. The final thermocycling conditions were as follows: an initial denaturation step of 5 min at 94 °C, followed by 35 cycles of 30 s of denaturation at 94 °C, 40 s of annealing (temperature dependent on the primer pairs, shown in Table 1), and 2.5 min of extension at 72 °C, with a final extension step at 72 °C for 7 min. Thermocycling of the ITS region were performed by modifying the protocol of White et al. (1990). The amplifications were carried out with an initial denaturation step of 2.5 min at 95 °C, followed by 30 cycles of 30 s of denaturation at 95 °C, 30 s of annealing at 56 °C, and 2 min of extension at 72 °C, with a final extension step at 72 °C for 10 min.
Amplified products were purified using a CleanSweep PCR Purification (Applied Biosystems, Waltham, MA, USA) kit according to the instructions provided by the manufacturer. Cleaned products were sequenced from both directions using an ABI PRISM 3100 Genetic Analyzer automated DNA sequencer.
Sequence Alignment and Phylogenetic Analysis
We used BioEdit 7.2.5 (Hall,1999) sequence alignment editor software to edit the sequence chromatograms, with visual inspection being performed for all polymorphic sites detected. Alignments were performed using ClustalW (Thompson et al.,1994) as implemented in BioEdit. The matrices of the five plastid sequences were concatenated into a single matrix using the SeaView 4.6.4 program (Gouy et al.,2010).
Phylogenetic relationships between the Hedera species were examined based on maximum likelihood (ML) and Bayesian approaches. ML analysis was performed using the RAxML-NG web server (http://embnet.vital-it.ch/raxml-bb/) (Kozlov et al.,2019) with 10 random starting trees and an autoMRE bootstrap search with 0.03 cut-off. The concatenated alignment was treated as a partitioned dataset. Evolutionary models were selected based on jModelTest results (Posada,2008). On the basis of Bayesian information criterion values, the F81 model was used for the five chloroplast regions, with Fatsia japonica and Travesia suindica being used as outgroups. Bayesian phylogenetic trees were reconstructed based on MrBayes v.3.2.7a (Ronquist et al.,2012) using the GTR+I+Γ model (general time-reversible model with a proportion of invariable sites and a gamma-shaped distribution of rates across sites) of sequence evolution. Four Markov chain Monte Carlo (MCMC) chains, one heated (temperature = 0.05) and three unheated, were run for 2,000,000 generations, sampling at every 1,000 generations. Information on gaps (indels) within the aligned sequence matrices were coded as binary characters. Gaps were coded following the simple indel coding algorithm (Simmons & Ochoterena,2001) using the program FastGap 1.2 (Borchsenius,2009).
PopART (Leigh & Bryant,2015) with implemented Templeton–Crandall–Singh (TCS) statistical parsimony network analysis (Clement et al.,2002) was used to evaluate genealogical relationships among sequences. Each insertion/deletion was considered to be a single mutation event, and all indels were therefore coded as single positions in the final alignments. The connection limit for the TCS analysis was 95% and gaps were treated as a fifth state.
Results
Sequence Variation
On the basis of sequence alignments of the selected five chloroplast markers, 40 new sequences were generated for the four examined Hedera taxa. The five concatenated chloroplast datasets of the 34 samples contained 3,783 base pairs after alignment, with 27 variable and 17 informative sites within Hedera (Table 2). As has previously been reported for chloroplast markers (Green et al.,2011; Grivet & Petit,2002; Valcárcel et al.,2003), we found that sequences showed a low degree of polymorphism, with a variability of only 0.7% being detected in plastid regions. Although originating from different geographical locations, we were unable to detect any variation in the chloroplast sequences of the different H. crebrescens specimens. At a microsatellite loci in the psbA-3′trnK-matk region, the H. crebrescens sequences had a species-specific insertion, which was not present in any of the other studied sequences. Hedera crebrescens had 12 T bases at this loci (7–11 T bases in other species). However, this was the only site that showed variability compared with those of the other species.
Compared with plastid regions, nuclear ITS sequences contained a proportionally greater number of polymorphic sites, showing 3.7% variability. Among all Hedera samples, we detected 22 variable and 14 informative sites. Hedera crebrescens was found to vary from the other three examined species with respect to two SNP loci. Moreover, of these two sites, one showed no variation among H. crebrescens samples, whereas the other site showed variation in one of the five samples.
Haplotype Network
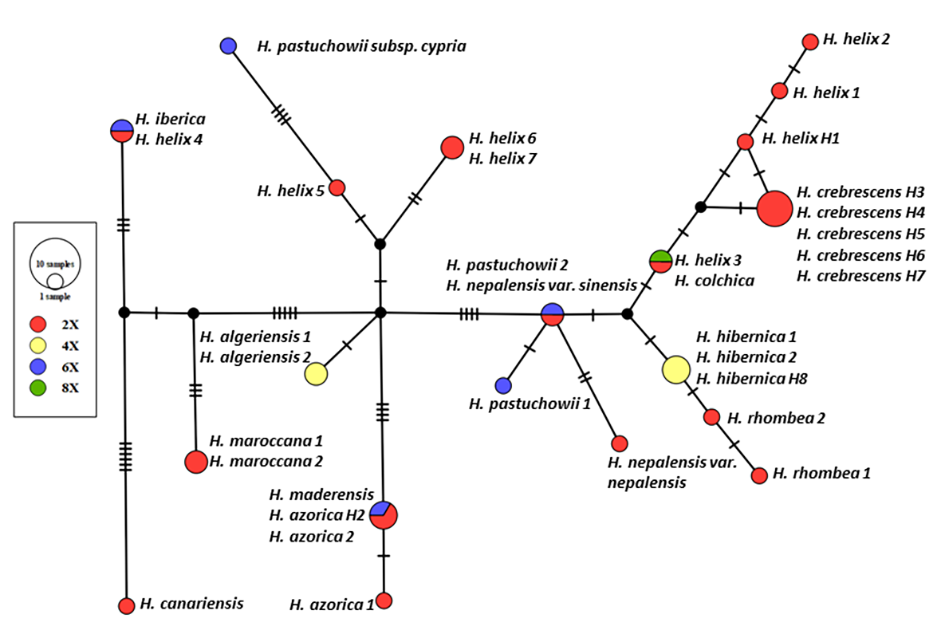
Unrooted haplotype genealogies were estimated based on the substitution polymorphisms (including indels coded as single characters) observed within the five plastid regions (Figure 2). TCS analysis of the 34 sequences of the 15 Hedera taxa yielded 20 haplotypes. The connection of haplotypes within the network followed a geographical structuring. In contrast, H. helix specimens were characterized by different haplotypes (eight specimens formed seven haplotypes) of different lineages. Each of the five H. crebrescens samples constituted an independent haplotype, which were placed at a single mutation step distance from the H. helix H1 specimen sampled in Hungary.
Figure 2
The haplotype network obtained based on Templeton–Crandall–Singh analysis of five concatenated cpDNA regions of Hedera samples. Black dots indicate missing intermediate haplotypes that were not observed in the analyzed sample set. Hash marks on branches represent mutation steps (number of base pair changes) between haplotypes. The different colors indicate the ploidy levels (red = 2x, yellow = 4x, blue = 6x, and green = 8x).
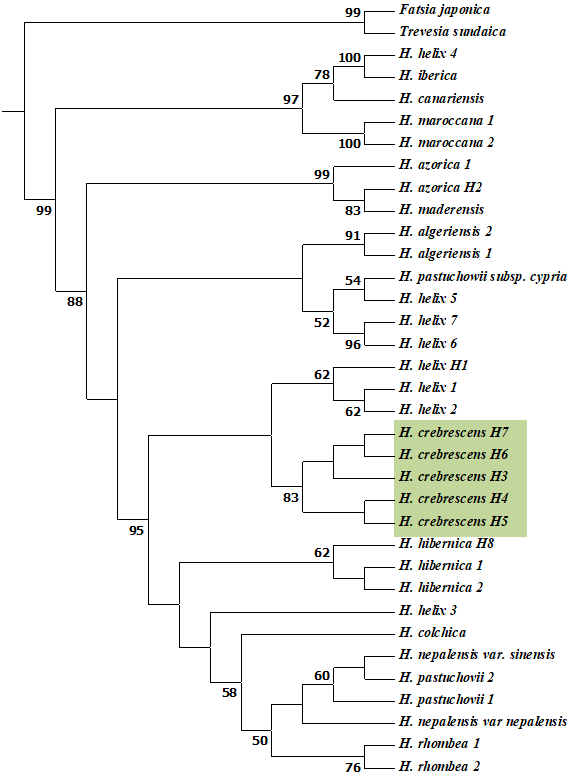
Chloroplast Phylogenetic Reconstruction
Phylogenetic analysis of the concatenated chloroplast matrix strongly supported the monophyly of Hedera (Figure 3, Figure S1). The strict consensus ML tree grouped Hedera sequences into two well-supported clades (99%), one of which included Western Mediterranean species (H. maroccana, H. canariensis, and H. iberica) and one sample of H. helix, whereas the other clade was divided into two clades, one of which separated two other Western Mediterranean taxa (H. azorica and H. maderensis) and the second contained the remainder of the taxa with weaker support. One clade contained the Western Mediterranean H. algeriensis, the Eastern Mediterranean H. pastuchowii subsp. cypria, and three H. helix specimens. The five H. crebrescens samples formed a single group and were connected to three other H. helix samples. The Western European H. Hibernica, the Asian species H. colchica, H. nepalensis, H. pastuchowii, and H. rhombea, a single H. helix specimen, were separated with high support from the clade containing H. crebrescens and H. helix samples.
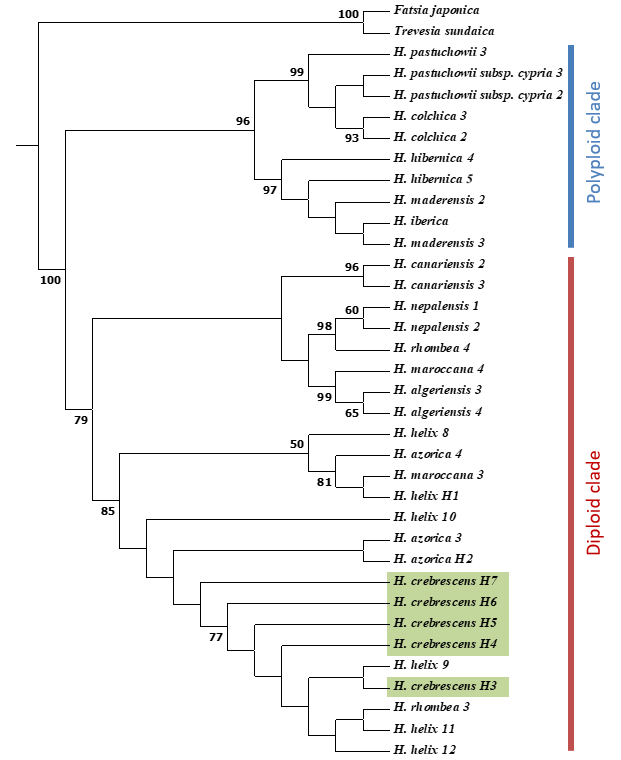
ITS Phylogenetic Reconstruction
Nuclear phylogeny also supported the monophyly of the genus and grouped taxa into two well-defined clades, one containing diploids and the other containing the polypoid taxa (Figure 4, Figure S2). Owing to the limited resolution of the diploid clade, as has been found in previous studies (Valcárcel et al.,2003,2017; Valcárcel & Vargas,2012; Vargas et al.,1999), we were unable to use ITS phylogeny as a basis for drawing clear conclusion regarding the phylogenetic position of H. crebrescens. However, nuclear phylogeny showing that H. crebrescens belongs to the diploid clade is consistent with the findings of previous cytological studies (Bényei-Himmer et al.,2017).
Discussion
Although several studies (Ackerfield & Wen,2003; Green et al.,2011; Grivet & Petit,2002; Valcárcel et al.,2003,2017; Valcárcel & Vargas,2012; Vargas et al.,1999) conducted in recent decades have examined the phylogeny of the genus Hedera, certain lineages have remained insufficiently well clarified. In some cases, species segregation has been based on low numbers of variable loci, whereas hybridization, polyploidization, rapid diversification, and recent evolutionary events may have further contributed to complicating the detection of molecular relationships within the genus (Ackerfield & Wen,2003; Green et al.,2011; Valcárcel et al.,2003,2014,2017; Vargas et al.,1999).
In the present study, we found that phylogeny based on plastid variation supported the monophyly of Hedera, although the phylogenetic trees did not show a strong and readily interpretable topology, and some subclades exhibited weak statistical support. Our consensus tree based on the analyses of five chloroplast regions showed a topology similar to that previously obtained based on phylogenetic cpDNA analyses (Green et al.,2011; Valcárcel et al.,2003). Previous molecular studies have established that, with the exception of H. helix, Mediterranean species are well separated from the Asian and European species. In contrast, overall phylogenetic relationships among European and Asian species, including H. crebrescens, tended to be poorly resolved due to the low statistical supports and limited resolution at lower levels. Hedera helix was found to be polyphyletic, as specimens were placed separately in the ML tree as well as in the haplotype network. This is consistent with the findings of previous studies that have revealed differences in lineages and chlorotypes within H. helix (Green et al.,2011; Grivet & Petit,2002; Valcárcel et al.,2003). The long-term cultivation of multiple species and cultivars can promote the mixing of genetic lineages owing to hybridization events and recent gene flow. On the basis of molecular evidence, the genus Hedera, including H. helix, has an Asian origin and there has been recent diversification in the Mediterranean region (Valcárcel et al.,2003,2017; Valcárcel & Vargas,2012). It is assumed that Central European taxa preserve imprints of the eastern genetic lineage, whereas H. helix has further extended its distribution toward the Mediterranean, where mixing of genetic lineages has occurred. Accordingly, the observed polyphyly of H. helix, due to the diversity of its genetic makeup, may cause uncertainties with respect to the molecular classification of the derived taxa. Although we found that the clade containing all European and Asian species had low support, all H. crebrescens specimens grouped together. On the basis of our analysis of five chloroplast regions, it appears that H. crebrescens is related to European populations of H. helix and H. hibernica and to certain Asian species (H. colchica, H. rhombea, H. pastuchowii, and H. nepalensis), However, on the basis of trichome morphology, which has previously been considered a taxonomically important trait for differentiating ivies, a close relationship with Asian taxa appears unlikely, due to the fact that unlike all Asian species, which have reddish scale-like trichomes, H. crebrescens has white stellate trichomes (Lum & Maze,1989; McAllister,1981; Rutherford et al.,1993). However, clarifying unequivocally the taxonomic position of H. crebrescens will clearly necessitate a more comprehensive survey of the entire Hedera genome. Nevertheless, the invariability we observed among H. crebrescens specimens of different geographical proveniences and its species-specific haplotype (unique loci that are not present in other species), provide support for its taxonomical autonomy. The previously hypothesized identity of H. crebrescens as H. hibernica (Udvardy & Bényei-Himmer,1999) can be refuted on the basis of molecular data, and also on previously obtained cytological results, as H. crebrescens is a diploid species. The closest relatives based on the chlorotypes were found to be in a lineage including H. helix specimens. However, the significant morphological, phenological, and ecological differences preclude identifying H. crebrescens as H. helix. Moreover, the leaves of these two taxa, on both vegetative and generative shoots, differ morphologically, and the reproductive organs also exhibit distinct characters (Bényei-Himmer et al.,2017). Hedera crebrescens is readily recognizable from its vigorous growth and large-lobed leaves on the emerging shoots, with dome-shaped apical lobe and overlapping leaf bases. The flattened fruits of this species are also characteristic, as they are tightly packed in the apical umbel of the inflorescence, which typically comprises a single rudimentary lateral umbel (Figure 1). The vigorous growth, high seed and fruit production, and a germination rate that is twice that of H. helix (Bényei-Himmer et al.,2017), are all conducive to the prolific spread of this species. Furthermore, its striking, dense, fleshy fruits prove to be particularly attractive to birds, highly contribute to the plant’s dispersal. Given this collection of favorable features, H. crebrescens has the potential to supplant H. helix in its native habitats. Moreover, the propensity for hybridization among Hedera species provides ample opportunities to introduce foreign genetic material into native populations. Indeed, hybridization in gardens, where large numbers of species and cultivars typically coexist, has previously been reported (Valcárcel et al.,2003), and might favor the selection of new lineages prone to garden escape.
By drawing attention to the newly described species H. crebrescens and highlighting its molecular background and relationships, we consider that the findings of this study provide a preliminary basis for forecasting the risks associated with the spread of this potentially invasive species in Central Europe. Identification and monitoring of this species will also be required in neighboring countries.
Supporting Material
The following supporting material is available for this article:
Figure S1. A consensus tree of the five concatenated cpDNA regions generated using the Bayesian method.
Figure S2. A consensus tree of the nrDNA ITS sequences generated using the Bayesian method.
Table S1. Location information for the sampled Hedera material.
Table S2. List of the sequences used in the study with respective location, voucher, and GenBank accession information.
Handling Editor
Agnieszka Popiela, University of Szczecin, Poland; https://orcid.org/0000-0001-9297-0538
Authors’ Contributions
EIM contributed in the sampling of the plant material, implemented the lab works, processed and evaluated the results, wrote the manuscript; EGT performed part of the molecular work and led the bioinformatics analyses; MB contributed in the conceptualization of the study, in the selection and sampling of the plant material; MH designed the study, evaluated the results, wrote the manuscript, supervised and financed the work
