. Introduction
Cinnamomum camphora is a vital woody resource for the extraction of natural linalool from essential oils. To achieve high yields and ensure sustainable utilization, a short-rotation coppice system is widely implemented in practical production. However, the coppice management system relies on frequent harvesting rotations, which inevitably leads to mechanical damage – a major abiotic stressor forplants (Taylor, 2023). In the case of plants, the stumps must not only prevent desiccation and pathogen infiltration through wound healing but also rapidly initiate the regeneration of adventitious buds (Ikeuchi et al., 2016; Omary et al., 2023). With the increase in Harvesting Cycles, stumps often exhibit a decline in sprouting vigor, delayed regeneration, or even mortality (Spinelli et al., 2017), which severely constrains the sustainable development of the industry. Therefore, elucidating the physiological response mechanisms of stumps following mechanical damage and identifying the key regulatory factors that maintain their regeneration potential remain critical biological questions to be addressed. In forestry practice, the application of wound-healing agents is a conventional measure to promote wound repair in trees. Previous studies have primarily focused on the role of wound dressings as physical barriers in reducing sap loss and blocking pathogens (Lonsdale, 1984). Although it has been reported that wound treatments can significantly enhance stump survival rates and sprout growth, the underlying molecular mechanisms driving this promotion remain to be elucidated.
Plant regeneration is a highly energy-intensive biological process that exhibits a “growth-defense trade-off” with the plant’s stress response (He et al., 2022). Upon mechanical wounding, damaged tissues rapidly synthesize and accumulate jasmonic acid (JA) within minutes, which modulates downstream stress-responsive pathways and is associated with the induction of resistance genes (Cunha et al., 2023; Li et al., 2022; Wasternack et al., 2006). While initiating this defensive response, the plant simultaneously suppresses vegetative growth to prioritize survival. However, for economic forests that require rapid biomass recovery, an excessive defensive response – manifested as hyper-elevated JA levels – may hinder sprout regeneration due to resource sequestration. Consequently, we hypothesize that wound-healing agents could facilitate regeneration by alleviating physiological stress and buffering the over-expression of JA, thereby optimizing internal energy allocation.
To test the aforementioned hypothesis, this study utilized 2-year-old Cinnamomum camphora ‘Nan’an No. 1’ stumps as research subjects. By integrating physiological and biochemical analyses with root transcriptomic (RNA-seq) technology, we compared the differential responses of stumps under natural healing versus wound treatment. Our research specifically focused on key gene families within the JA biosynthetic pathway (LOX, AOS, AOC, OPR, and ACX) and related signaling networks, such as the MAPK cascade. This study aims to elucidate, at the molecular level, the mechanisms by which wound management promotes sprouting regeneration by modulating hormonal signaling balance, thereby providing a theoretical basis for post-harvest physiological regulation and targeted cultivation of subtropical aromatic oil-bearing forests.
. Materials and methods
. Plant materials and experimental design
The experimental site is situated at the C. camphora Plantation Base of Quanzhou Mingdao Agricultural and Forestry Development Co., Ltd. in Xiangyang Township, Nan’an City, Fujian Province (24° 34' N, 118° 08' E and altitude ranging from 890 m to 1000 m). The area features a subtropical monsoon climate, with an annual average temperature of 20°C and an annual average rainfall of 1760 mm. The experimental site consisted of two-year-old Cinnamomum camphora ‘Nan’an No. 1’ clones with uniform growth. These plants were transplanted in 2018 and had not been subjected to any prior harvesting measures before the study.
This study utilized a randomized complete block design with three replicates per treatment group. Three distinct experimental groups were established: (1) the control group (CK), consisting of unharvested plants to represent the baseline physiological state; (2) the natural healing group (BT), in which plants were decapitated (coppiced) at approximately 20 cm above the ground, with the resulting stump wounds left untreated to heal naturally; and (3) the wound dressing treatment group (T), which underwent the same decapitation procedure but received a paste-like wound sealant (Guoerguang-Hutuhu; Sichuan Guoguang Agrochemical Co., Ltd., China) applied immediately after cutting to completely cover the exposed surfaces.
. Sample collection
Fine root samples were collected from each treatment group to analyze the physiological and molecular responses to harvesting stress. For physiological analysis, sampling was conducted on the day of harvesting (Day 0, as the CK baseline) and on Days 5, 15, and 30 post-harvesting (labeled as D5, D15, and D30, respectively). Based on preliminary observations indicating that Day 5 represents a critical window for stress response, root samples from the CK (Day 0), T-D5, and BT-D5 groups were specifically selected for transcriptomic sequencing (RNA-seq). All samples were immediately rinsed with sterile water, flash-frozen in liquid nitrogen, and stored at –80°C for subsequent analysis.
. Assessment of physiological indicators and sprout regeneration
In March 2022, the regeneration capacity of the stumps was evaluated by measuring the length, ground diameter, and biomass of the newly emerged sprouts. For physiological assessments, the endogenous jasmonic acid (JA) content in the roots was quantified using liquid chromatography-mass spectrometry (LC-MS) (Pan et al., 2010). The activities of superoxide dismutase (SOD) and peroxidase (POD) were determined using the nitroblue tetrazolium (NBT) photoreduction method and the guaiacol method, respectively. Three biological replicates were established for each time point.
. RNA extraction, library construction, and sequencing
Total RNA was extracted from the root samples (CK, T-D5, and BT-D5) using the RNAprep Pure Plant Plus Kit (Polysaccharides & Polyphenols-rich; TIANGEN Biotech, Beijing, China) following the manufacturer’s instructions. Prior to library construction, the integrity and concentration of the isolated RNA were verified. The cDNA libraries were constructed and sequenced on the BGISEQ-500 platform (BGI-Shenzhen, Shenzhen, China), generating 100 bp paired-end reads.
. Transcriptomic analysis and identification of JA-related genes
Raw reads were filtered to obtain clean reads, which were then mapped to the C. camphora reference genome using Bowtie2. Gene expression levels were quantified using RSEM and normalized as Fragments Per Kilobase of transcript per Million mapped reads (FPKM). Differentially Expressed Genes (DEGs) were identified using DESeq2 with a threshold of |log2(Fold Change)| ≥ 1 and a False Discovery Rate (FDR) < 0.05. Functional annotation was performed using BLAST against the NR, Swiss-Prot, GO, KEGG, and PlantTFDB databases. To identify key genes in the JA biosynthesis pathway, Hidden Markov Model (HMM) profiles for the LOX (PF00305) and OPR (PF00724) domains were used to search the C. camphora protein dataset. Other genes (AOS, AOC, ACX) were identified by BLASTP using Arabidopsis thaliana homologs as queries. Phylogenetic trees were constructed using MEGA 6.0 via the Neighbor-Joining (NJ) method.
. Quantitative real-time PCR (qRT-PCR) validation
To validate the RNA-seq data, four DEGs associated with hormone and MAPK signaling pathways were selected for qRT-PCR. Primers were designed using Primer 5.0 (Table 1), qRT-PCR was performed using the BrightCycle Universal SYBR Green qPCR Mix on a fluorescence quantitative PCR instrument. The reaction program was 37°C for 2 minutes, 95°C for 3 minutes, 95°C for 5 seconds, 60°C for 30 seconds, with 40 cycles. Relative expression levels were calculated using the 2-ΔΔCTmethod (Tang et al., 2024), with Actin serving as the internal reference gene.
Table 1
qRT‑PCR validation primer sequences for transcriptome data.
. Identification and bioinformatics analysis of JA-related gene families
The genome annotation information, protein sequence, and coding sequence data files of C. camphora were downloaded through the National Genomics Data Center (NGDC) (Tang et al., 2024). The genome data of Arabidopsis thaliana were downloaded through the TAIR database of A. thaliana (https://www.arabidopsis.org/). The genome data of upland cotton were downloaded through NCBI (https://www.ncbi.nlm.nih.gov/). The Hidden Markov Model files (PF00305, PF00724) containing LOX conserved domain information and OPR conserved domain information were downloaded from the Pfam database (http://pfam.xfam.org/). The Simple HMM Search function of TBtools was used to extract LOX and OPR genes. The candidate genes obtained by screening out duplicates with Excel software were further confirmed on the NCBI website’s Batch CD-search function (https://www.ncbi.nlm.nih.gov/Structure/bwrpsb/bwrpsb.cgi) to determine whether they contain a complete domain, and the gene sequences of LOX and OPR of C. camphora were obtained. For the remaining genes, taking the relevant genes of A. thaliana as the reference protein sequence, local blastP search and extraction were performed using local blast v3 (http://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/). The candidate genes obtained by screening out duplicates were further confirmed to determine whether they contain a complete domain, and the gene sequences of AOS, AOC, and ACX of C. camphora were obtained. Referring to the naming method of key genes identified in other species, “CcLOX”, “CcAOS”, “CcAOC”, “CcOPR”, and “CcACX” were used to abbreviate the IDs of key genes for JA synthesis in the genome of C. camphora. Chromosome localization and naming were performed using TBtools. The online tool MEME Motif Discovery (http://meme-suite.org/tools/meme) was selected to analyze the conserved motifs of key genes for JA synthesis in C. camphora to determine the characteristics of their conserved domains. For the CcLOX, CcAOC, and CcOPR genes, the maximum number of reference motifs was set to 10, and the other settings were left as default; for the CcACX gene, the maximum number of motifs was set to 18, and the other settings were left as default. Finally, the obtained motif information was visualized by TBtools software. The protein sequences of related genes of C. camphora, A. thaliana, and upland cotton were subjected to multiple alignments through MEGA6.0, and a phylogenetic tree was constructed using the NJ method and tested by Clustal X.
. Results
. Effects of wound management on sprout regeneration and physiological responses in Cinnamomum camphora
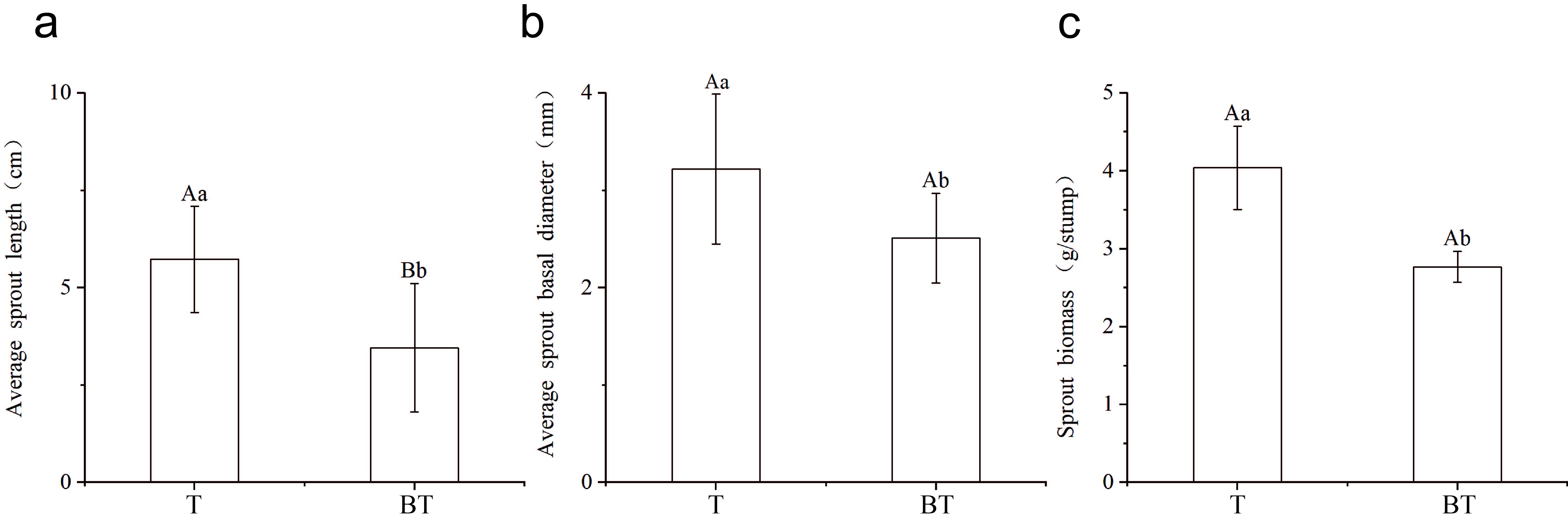
The results showed that wound management significantly improved the regeneration performance of Cinnamomum camphora stumps (Figure 1). In Treatment T, the sprout length reached 5.73 cm, representing a highly significant increase of 62.78% compared to the BT group (P < 0.01). Additionally, the average sprout ground diameter (3.21 mm) and sprout biomass (4.04 g/plant) in the T group were significantly higher than those in the BT group (2.53 mm and 2.76 g/plant, respectively; P < 0.05). This synchronized growth in sprout length, diameter, and biomass reflects a comprehensive enhancement of stump regeneration capacity following wound management.
Figure 1
Effect of wound management on budding regeneration in C. camphora.
Note: Different lowercase letters indicate significant differences between treatments (P < 0.05), while different uppercase letters indicate extremely significant differences between treatments (P < 0.01).
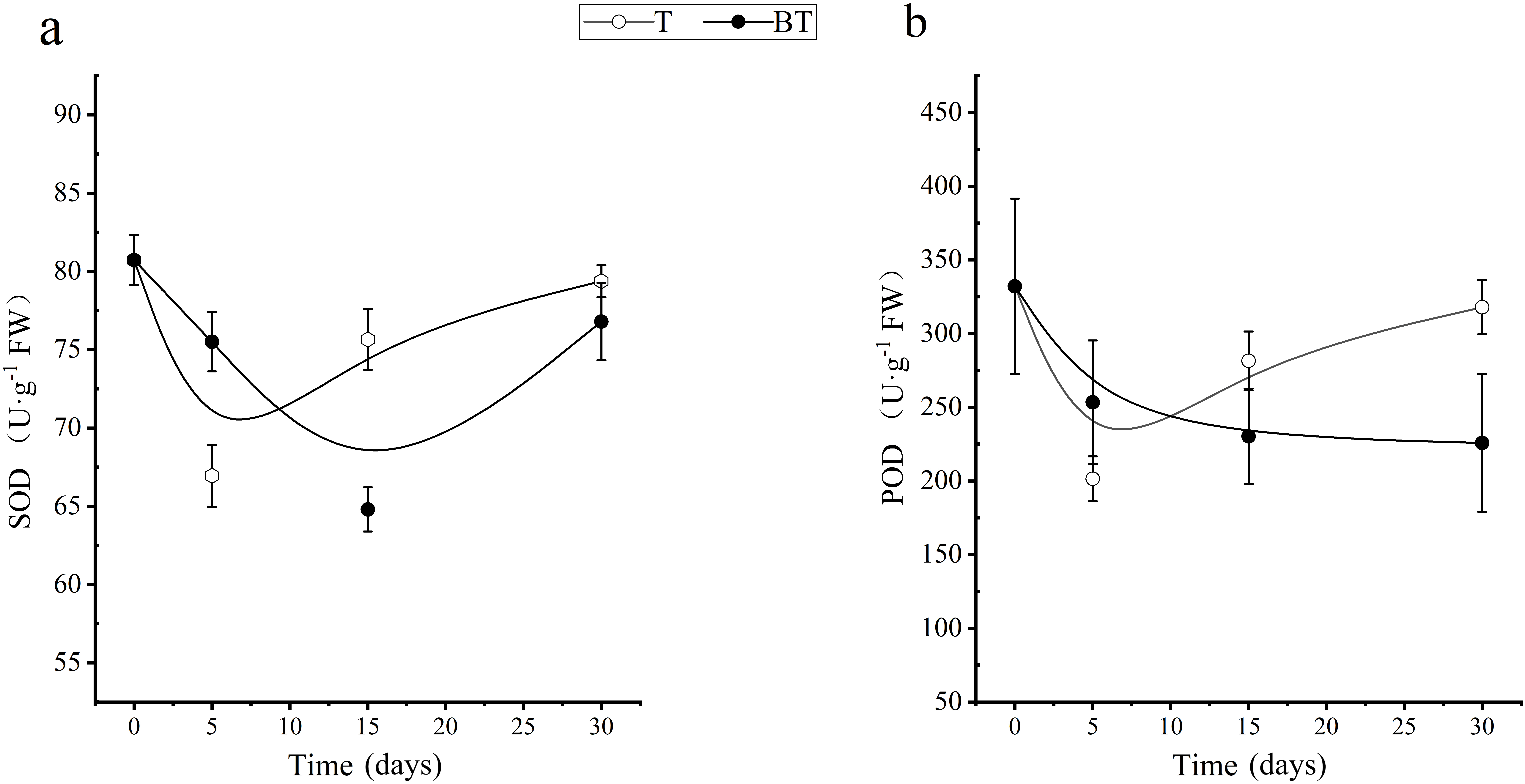
The results indicated that Treatment T significantly altered the antioxidant enzyme response patterns in roots under harvesting stress (Figure 2). Both SOD and POD activities in the T group exhibited a trend of initial inhibition followed by rapid recovery, with both reaching their lowest levels on Day 5 post-harvesting (67.01 and 201.24 U·g-1 FW, respectively). Subsequently, these activities increased sharply; by Day 30, they had recovered to 98.33% and 95.73% of their pre-harvesting baseline levels, respectively, effectively re-establishing physiological homeostasis. In contrast, physiological recovery in the BT group lagged significantly. Its SOD activity did not reach its minimum level (64.82 U·g-1 FW) until Day 15, while POD activity exhibited a continuous downward trend throughout the observation period, remaining significantly lower (a 32.00% reduction) than the baseline level by Day 30. These divergent antioxidant enzyme patterns correlate consistently with the sprout growth vigor observed in Figure 1. Specifically, the earlier rebound of enzyme activities in the T group was accompanied by superior regeneration capacity, whereas the sustained suppression of enzyme activities in the BT group corresponded to weaker growth performance.
Figure 2
Changes in root SOD and POD activities under different wound treatments.
Note: T: applied wound healing agent; BT: no wound healing agent applied. The same below.
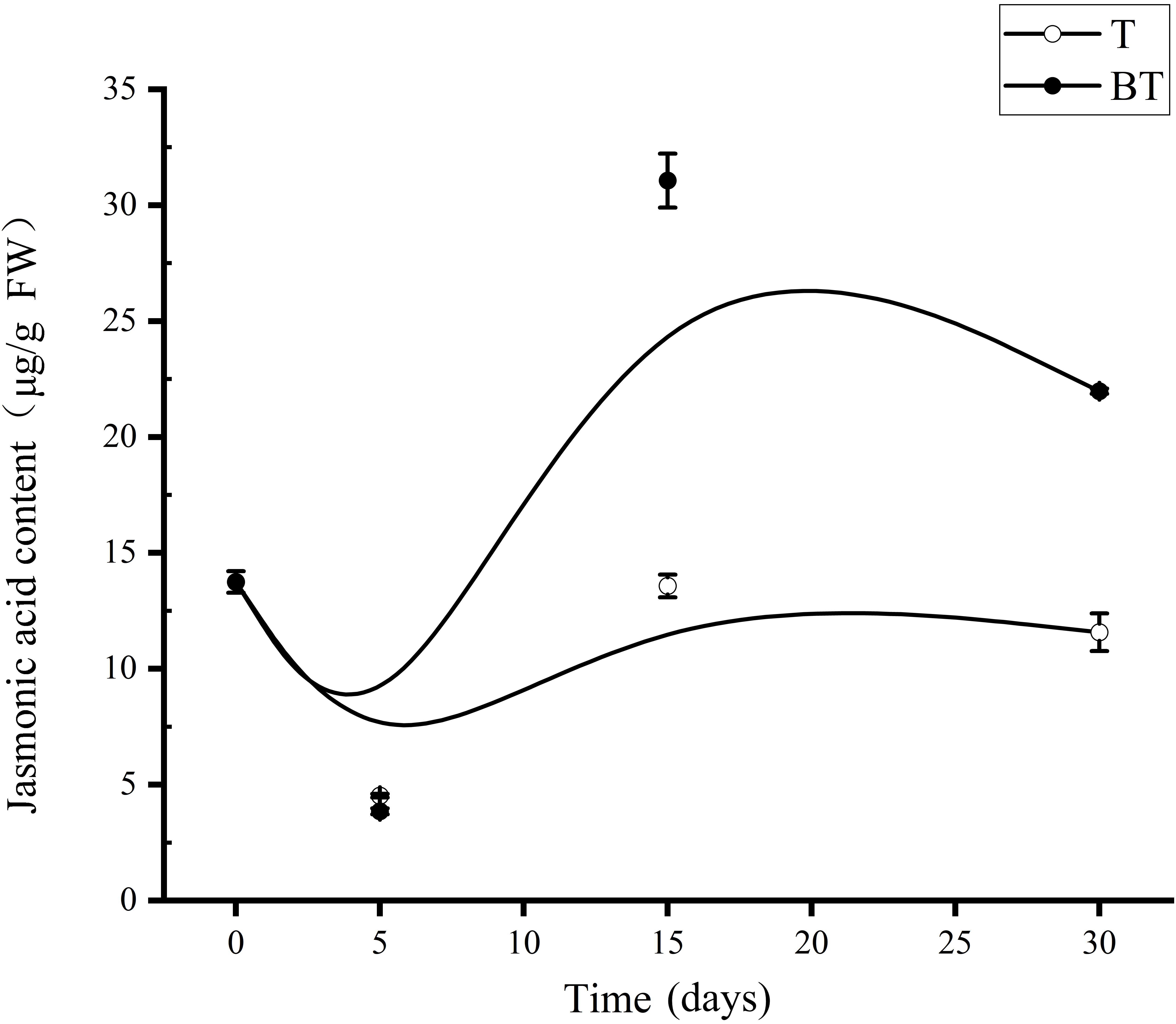
The results of endogenous jasmonic acid (JA) content indicated that the BT group exhibited more pronounced dynamic fluctuations in JA levels compared to the T group (Figure 3). During the temporal response following harvesting, JA concentrations in both groups reached their nadir on Day 5, with values recorded at 4.53 ± 0.08 μg/g FW in the T group and 3.85 ± 0.13 μg/g FW in the BT group. Subsequently, JA levels in both groups peaked on Day 15. However, the peak concentration in the BT group reached 31.06 ± 1.17 μg/g FW, which was 2.26–fold higher than its initial level (13.75 μg/g FW) and highly significantly greater than that of the T group at the same time point (13.58 ± 0.49 μg/g FW; P < 0.01). By Day 30 post-harvesting, JA levels in the T group had preemptively declined to 11.58 ± 0.81 μg/g FW, essentially returning to pre-harvesting levels. In contrast, the BT group maintained a significantly higher concentration (21.98 ± 0.11 μg/g FW). These response characteristics of JA content correlate with the growth performance shown in Figure 1. Specifically, the pronounced JA peaks observed in the BT group are consistent with the restricted sprout regeneration, whereas the relatively stable JA fluctuations in the T group were accompanied by superior regenerative vigor.
. Root transcriptomic profiling and functional annotation
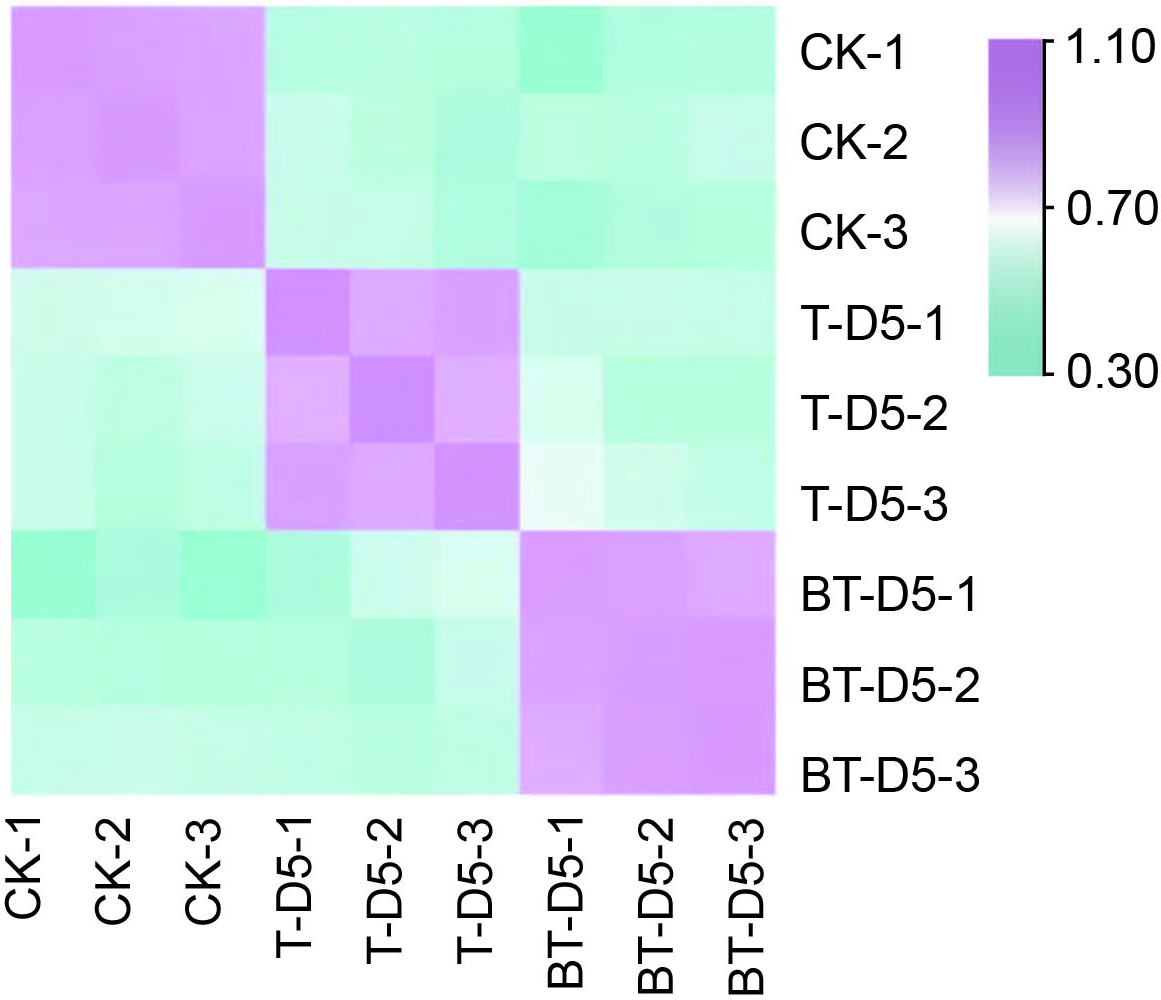
Transcriptome sequencing and filtering were carried out on the roots of C. camphora before logging (CK), on the 5th day after logging without the application of the wound-healing agent (BT-D5), and on the 5th day after logging with the application of the wound-healing agent (T-D5). The results obtained are shown in Table 2. The total number of Q20 and Q30 reads of all sequenced samples is greater than 98%, and the ratio of clean reads mapped to the genome is greater than 81.24%, indicating that the quality of the transcriptome sequencing meets the requirements and can be used for assembly and expression analysis. The correlation analysis of the gene expression between samples (Figure 4) revealed that the correlation of the repeated samples in each of the CK, T-D5, and BT-D5 treatments is greater than 99%, indicating that the repeatability of the samples within each treatment is relatively good.
Table 2
Reads quality statistics.
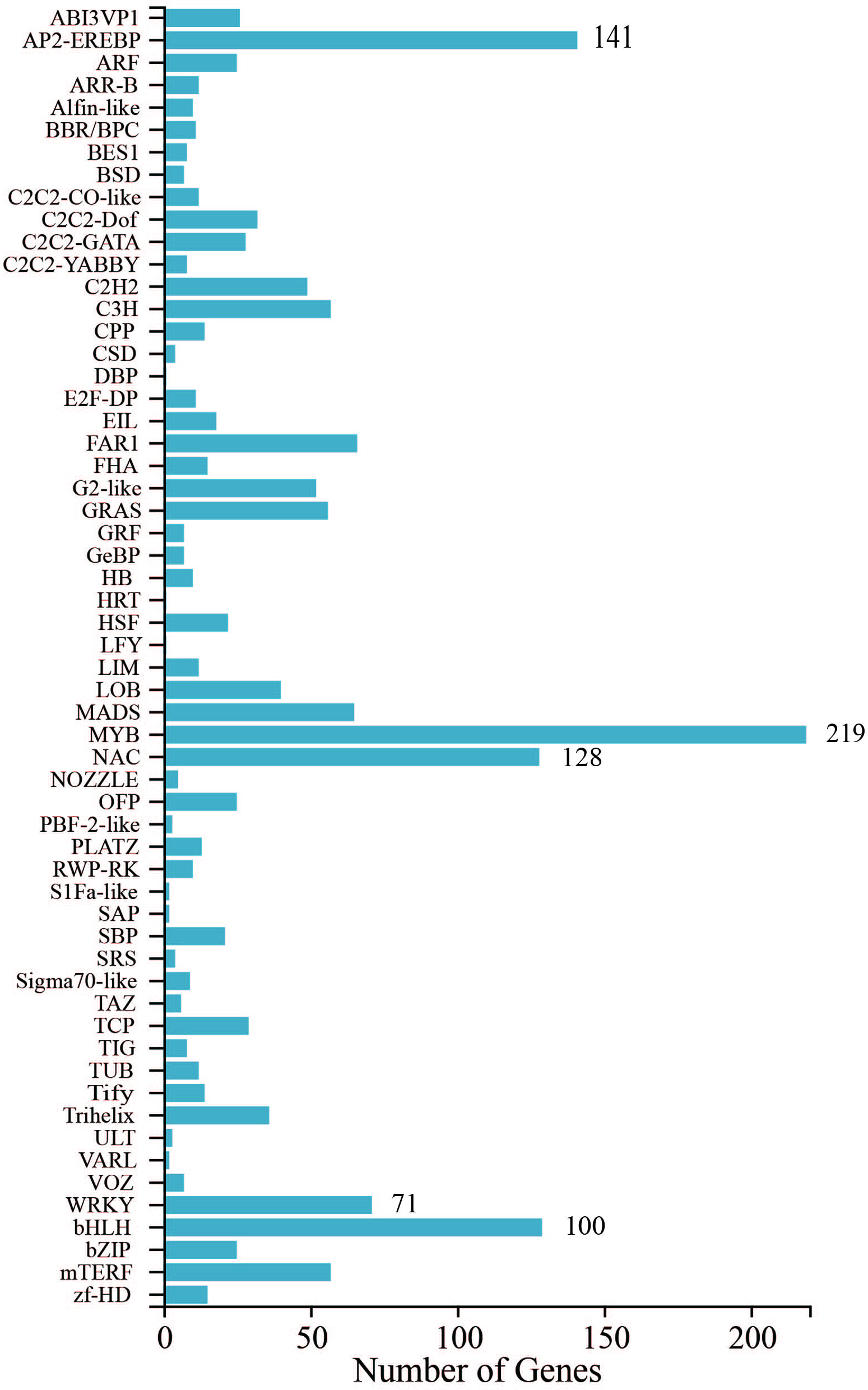
Furthermore, TF annotation was performed on 1638 genes identified in 9 samples (Figure 5), with the MYB family having the highest number of annotated genes, totaling 219. Next are the AP2-EREBP family (141), the NAC family (128), the bHLH family (100), and the WRKY family (71). These transcription factor (TF) families play pivotal roles in plant stress responses and wound repair signaling transduction. Consequently, they provide a valuable reference for the subsequent screening of core differentially expressed genes (DEGs) associated with wound dressing treatments.
. Identification and functional enrichment of DEGs
. Comparative analysis of differentially expressed genes
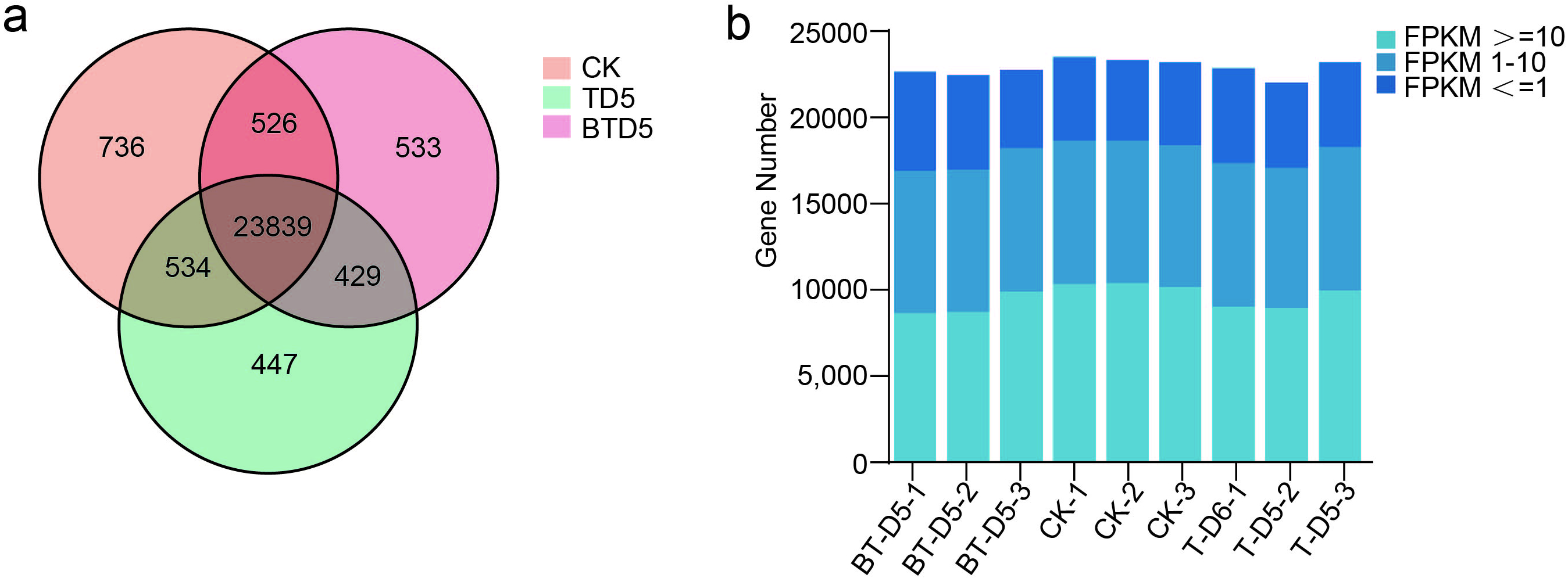
A total of 27,053 genes were identified across the nine transcriptome sequencing samples (Figure 6a). Among these, 23,839 genes were shared by the CK, BT-D5, and T-D5 groups, accounting for 88.12% of the total genes. Furthermore, the analysis revealed 736, 533, and 447 group-specific genes in the CK, BT-D5, and T-D5 groups, respectively. The number of genes shared between CK and T-D5 (543) was slightly higher than that between CK and BT-D5 (526). The analysis of the Fragments Per Kilobase of transcript per Million mapped reads (FPKM) distribution revealed significant differences in expression abundance among samples (Figure 6b). In the low-expression interval (FPKM ≤ 1), the T-D5 group exhibited the lowest proportion of genes. Conversely, in the medium (1 < FPKM ≤ 10) and high (FPKM & 10) expression intervals, the proportion of genes in the BT-D5 group was significantly lower than those in both the CK and T-D5 groups. Statistical results indicated that the overall gene expression abundance in roots decreased following harvesting compared to the CK group; however, across all expression gradients, the gene proportions in the T-D5 group remained consistently higher than those in the BT-D5 group.
. GO enrichment analysis of differentially expressed genes
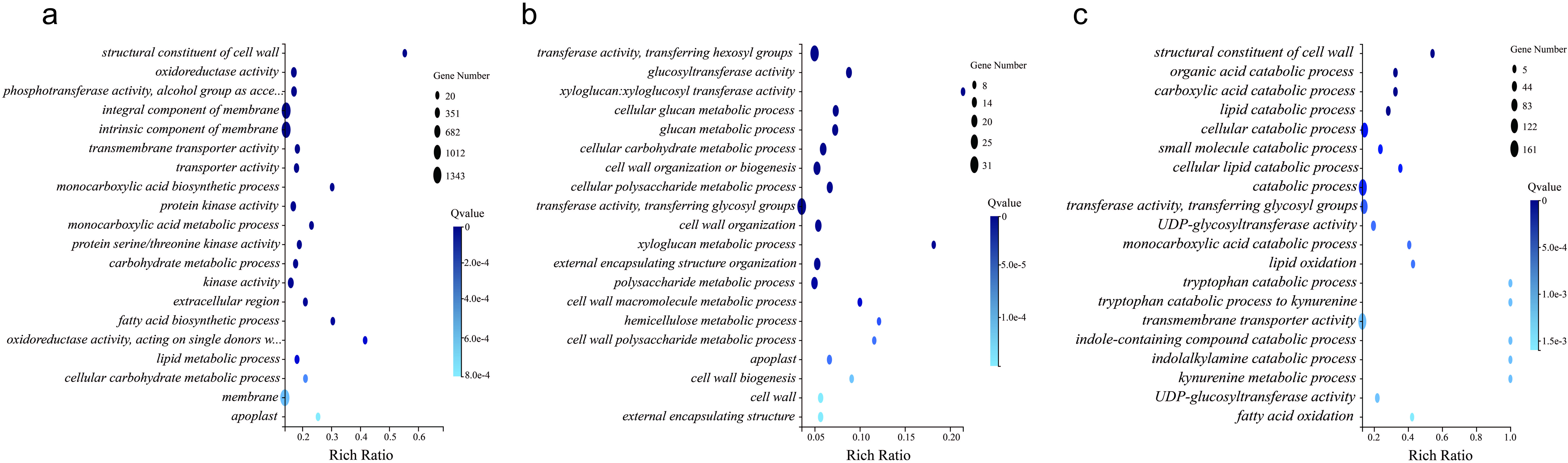
The GO functional enrichment analysis of differentially expressed genes (DEGs) in the CK-vs.-BT-D5, CK-vs.-T-D5, and T-D5-vs.-BT-D5 comparisons (Figure 7) revealed the divergent molecular response characteristics of roots under different treatments. The results showed that DEGs in the CK-vs.-BT-D5 comparison (Figure 7a) were significantly enriched in the membrane and membrane-associated component terms. For the CK-vs.-T-D5 comparison (Figure 7b), DEGs were primarily enriched in catabolic processes and transmembrane transporter activities. Notably, DEGs in the T-D5-vs.-BT-D5 comparison (Figure 7c) exhibited high enrichment in transferase activity, transferase-related groups, and processes associated with cell wall organization or biosynthesis. These findings suggest that the functional differences between the T-D5 and BT-D5 groups are primarily concentrated in the synthesis and remodeling pathways of cell wall components.
. KEGG pathway enrichment analysis of DEGs
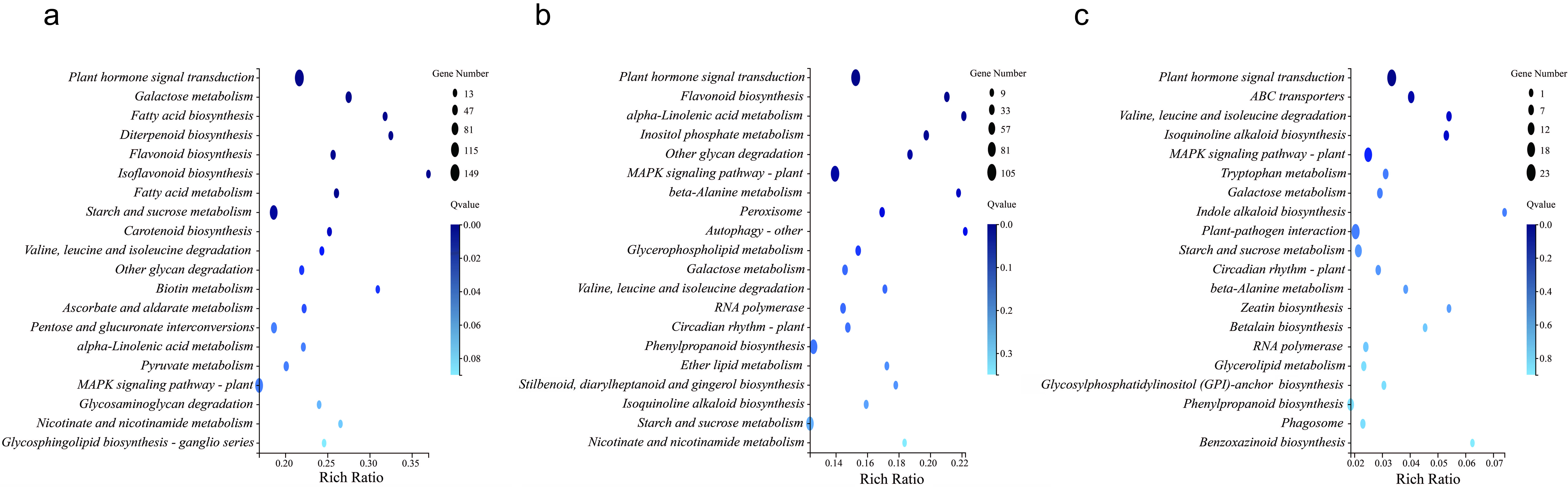
The KEGG functional enrichment analysis was performed on the differentially expressed genes (DEGs) of CK-vs.-T-D5, CK-vs.-BT-D5, and T-D5-vs.-BT-D5 (Figure 8). The DEGs of CK-vs.-BT-D5 (Figure 8a) are significantly enriched mostly in plant hormone signal transduction, the plant MAPK signaling pathway, and starch and sucrose metabolism. The DEGs of CK-vs.-T-D5 (Figure 8b) are significantly enriched mostly in plant hormone signal transduction, the plant MAPK signaling pathway, and phenylpropanoid biosynthesis. The DEGs of T-D5-vs.-BT-D5 (Figure 8c) are significantly enriched mostly in plant hormone signal transduction, the plant MAPK signaling pathway, and plant-pathogen interaction. It can be seen that plant hormone signal transduction and the plant MAPK signaling pathway are important pathways for the differential gene expression in C. camphora before and after logging.
. qRT PCR validation of transcriptome sequencing
To further verify the accuracy of the transcriptome data, two significantly differentially expressed genes (Maker00007883, Maker00025064 genes, and Maker00005324, Maker00013322 genes) were selected from the plant hormone signal transduction and plant MAPK signaling pathways, respectively, for qRT-PCR verification (Figure 9). The results are consistent with the trend of the FPKM values, indicating that the sequencing data of the C. camphora transcriptome are accurate and highly reliable. These results provide a solid data foundation for the subsequent functional mining of differentially expressed genes.
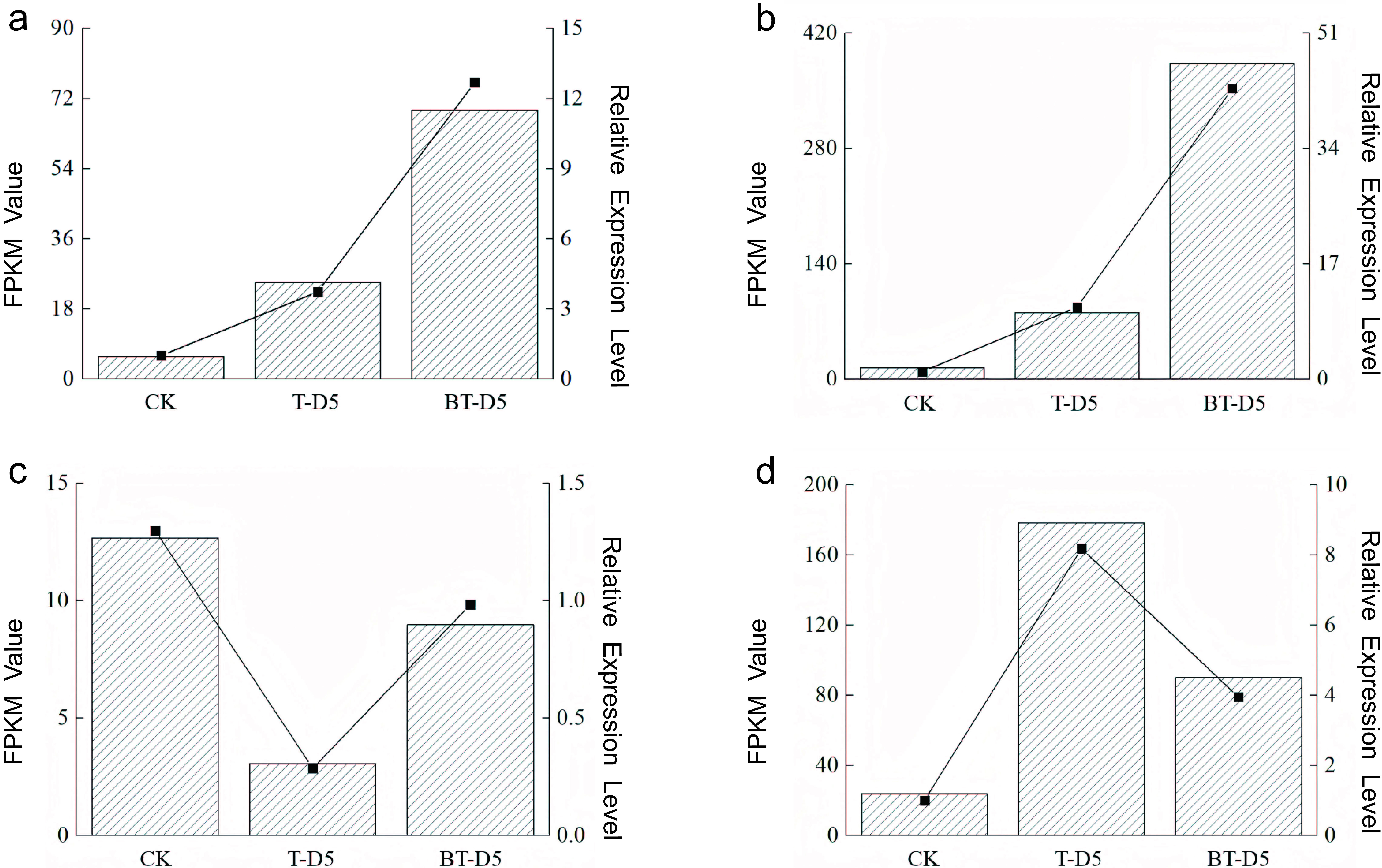
. Identification and characterization of the JA biosynthetic gene family
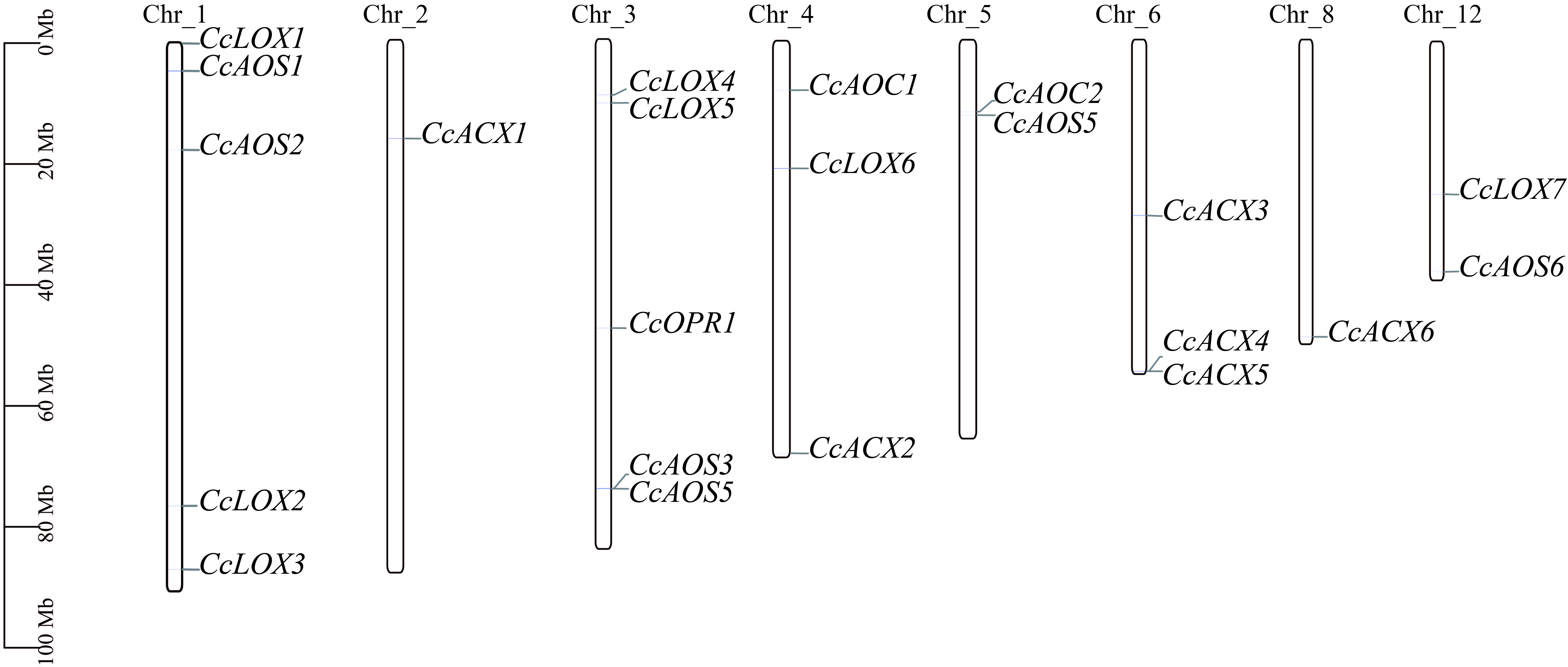
. Identification of candidate genes for JA biosynthesis
Seven LOX genes with complete identified domains were obtained in the local protein database of C. camphora through HMM Search. According to the chromosomal positions and order, they were named CcLOX1 to CcLOX7 (Figure 10). The protein length ranges from 821 to 1802 amino acids (aa), the molecular mass ranges from 93.73 to 202.31 kilodaltons (kDa), and the isoelectric point (PI) ranges from 5.46 to 7.85. The instability coefficient of CcLOX5 is lower than 40, which is a stable protein, and the rest of the LOX proteins are all unstable proteins. Six AOS genes were named CcAOS1 to CcAOS6. The protein length ranges from 142 to 158 aa, the molecular mass ranges from 5.76 to 133.55 kDa, and the PI ranges from 7.96 to 9.32. The instability coefficients of CcAOS5 and CcAOS6 are lower than 40, which are stable proteins, and the rest are all unstable proteins. Two AOC genes of C. camphora were named CcAOC1 and CcAOC2. The protein length ranges from 247 to 253 aa, the molecular mass ranges from 27.69 to 27.21 kDa, and the PI ranges from 8.99 to 9.38. The instability coefficients are all higher than 40, which are unstable proteins. One OPR gene (CcOPR1) was identified: the protein length is 397 aa, the molecular mass is 43.92 kDa, and the PI value is 6.55. The instability coefficient is 32.5, which is a stable protein. Six ACX genes of C. camphora were named CcACX1 to CcACX6. The protein length is 135 to 1284 aa, the molecular mass ranges from 14.39 to 142.53 kDa, and the PI ranges from 6.08 to 9.57. The instability coefficients of CcACX3, CcACX4, and CcACX6 are lower than 40, which are stable proteins, and the rest are all unstable proteins.
In the prediction results of subcellular localization, CcLOX1, CcLOX2, CcLOX3, CcLOX4, and CcLOX6 are located in the chloroplast, and CcLOX5 is located in the cytoplasm; CcAOS2 and CcAOS5 are located in the chloroplast, and the rest of the AOS proteins are located in the endoplasmic reticulum. The CcAOCs proteins are all located in the chloroplast, CcOPR1 is located in the peroxisome, and the CcACXs genes are all located in the peroxisome.
. Analysis of conserved motifs
The results of the similarity and diversity of the LOX proteins of C. camphora are shown in Figure 11a. A total of 10 different conserved motifs were identified and named motif 1 to 10. The LOX proteins of C. camphora all contain highly conserved motifs. Among them, the amino acid length of CcLOX6 is twice that of the other proteins. The conserved motifs were repeated. CcLOX1 repeats motif 5, CcLOX5 repeats motif 1, and CcLOX4 repeats motif 6.
A total of 10 different conserved motifs were identified in the AOS proteins of C. camphora (Figure 11b). Among them, motif 8, motif 2, and motif 7 form the conserved domain of the AOS proteins. The protein sequence of CcAOS5 is too short and only contains motif 5. CcAOS1, CcAOS3, and CcAOS4 all lack motif 5 and have motif 1, motif 3, motif 4, motif 6, and motif 9 that are not contained in other sequences. The sequences of CcAOS1 and CcAOS3 are relatively long and there are obvious fragment repeat motifs.
The sequences of C. camphora AOC were combined with those of Arabidopsis thaliana AOC for identification. The results are shown in Figure 11c. A total of 8 different conserved motifs were identified in the AOC proteins of C. camphora. The AOC proteins of C. camphora all contain highly conserved motifs. Compared with the AOC protein motifs of A. thaliana, motif 5 and motif 8 are missing, and motif 9 and motif 10 are added.
The motifs of the OPR protein of C. camphora and the OPR protein of A. thaliana were combined for identification. The results are shown in Figure 11d, and 9 different conserved motifs were identified. The CcOPR1 protein has highly conserved motifs. It is similar to the AtOPR3 of A. thaliana and lacks motif 10 compared with AtOPR1 and AtOPR2.
A total of 18 different motifs were identified in the ACX proteins of C. camphora (FFigure 11e). Except that CcACX5 may be due to an incomplete sequence, the rest of the ACX proteins all contain highly conserved motifs. Motif 4 (except for CcACX1), motif 5 (except for CcACX1 and CcACX3), motif 1, and motif 3 are all conserved in the ACX proteins of C. camphora. CcACX4 lacks motif 6.

. Phylogenetic analysis of gene families in the JA biosynthetic pathway
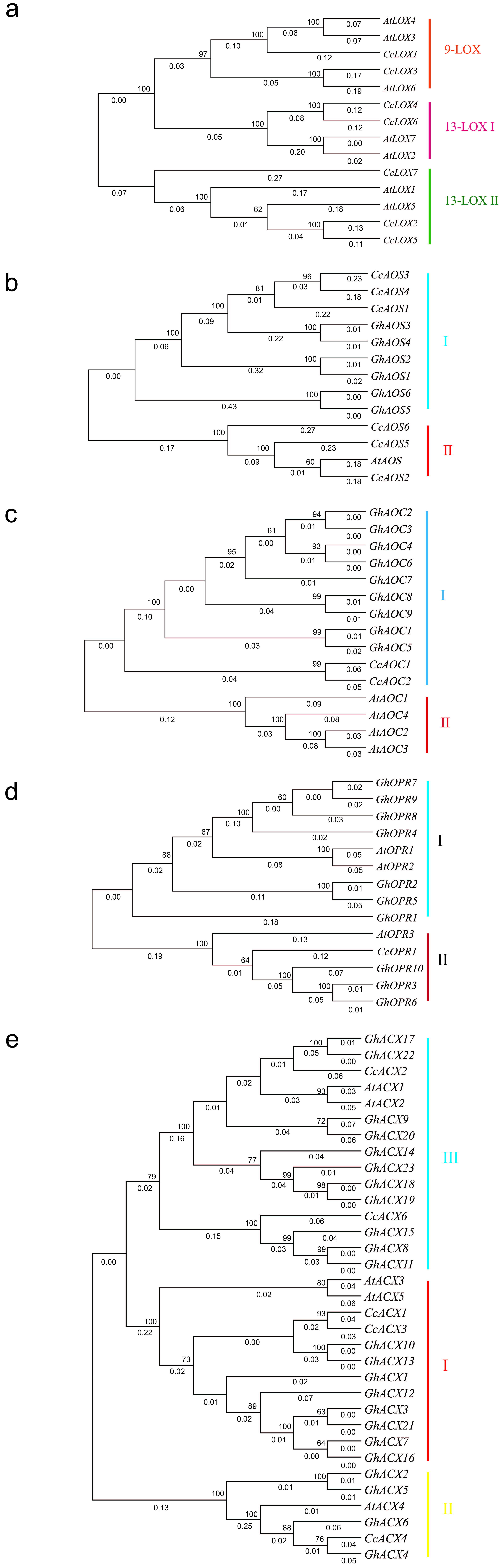
A phylogenetic tree was constructed using the conserved protein segment sequences of A. thaliana LOX and C. camphora LOX (Figure 12a). Based on the classification method of the A. thaliana LOX gene as a model plant, the CcLOX gene family of C. camphora is divided into two subfamilies: 9-LOX and 13-LOX. In the 9-LOX subfamily, CcLOX1 is closely related to AtLOX3 and AtLOX4, and CcLOX3 is closely related to AtLOX6. In 13-LOX, it is divided into 13-LOX type I and 13-LOX type II. CcLOX4 and CcLOX6 belong to 13-LOX type I and belong to the same type I as AtLOX2 and AtLOX7. CcLOX2, CcLOX5, and CcLOX7 belong to 13-LOX type II and belong to the same type II as AtLOX1 and AtLOX5.
A phylogenetic tree was constructed using the protein sequences of A. thaliana AOS, upland cotton AOS, and C. camphora AOS. The results are shown in Figure 12b. The AOS genes can be clearly clustered into 2 groups. The AOS genes of upland cotton are clustered in branch I. The AOS genes of A. thaliana are distributed in branch II. In turn, the AOS genes of C. camphora are distributed in both types I and II. It is likely that the AOS genes of C. camphora have undergone functional changes during the evolution process. CcAOS1, CcAOS3, and CcAOS4 have similar functions to the AOS genes of upland cotton, and CcAOS2, CcAOS5, and CcAOS6 have similar functions to the AOS genes of A. thaliana.
The phylogenetic results of the protein sequences of A. thaliana AOC, upland cotton AOC, and C. camphora AOC are shown in Figure 12c. The AOC genes can be clearly clustered into 2 groups. The AOC genes of upland cotton and C. camphora are clustered in Class I, while the AOC genes of A. thaliana are clustered in Class II.
The phylogenetic results of the protein sequences of A. thaliana, upland cotton, and C. camphora OPR (Figure 12d) show that the OPR genes can be clearly clustered into 2 groups. CcOPR1 is clustered in group II together with GhOPR3, GhOPR6, GhOPR10, and AtOPR3, and the rest of the OPRs are clustered into one group.
The results of the phylogenetic tree of the protein sequences of A. thaliana, upland cotton, and C. camphora ACX (Figure 12e) show that the ACX genes can be clearly clustered into 3 groups. Each group has 2 ACX genes of C. camphora, and they are all clustered together with the ACX genes of upland cotton, indicating that the evolutionary relationship between C. camphora and upland cotton is closer than that with A. thaliana.
Figure 12
Phylogenetic analysis.
Note: Figure a shows the evolutionary relationship of the LOX genes between C. camphora and A. thaliana; Figure b shows the evolutionary relationship of the AOS genes among C. camphora, Gossypium hirsutum, and A. thaliana; Figure c shows the evolutionary relationship of the AOC genes among C. camphora, G. hirsutum, and A. thaliana; Figure d shows the evolutionary relationship of the OPR genes among C. camphora, G. hirsutum, and A. thaliana; Figure e shows the evolutionary relationship of the ACX genes among C. camphora, G. hirsutum, and A. thaliana.
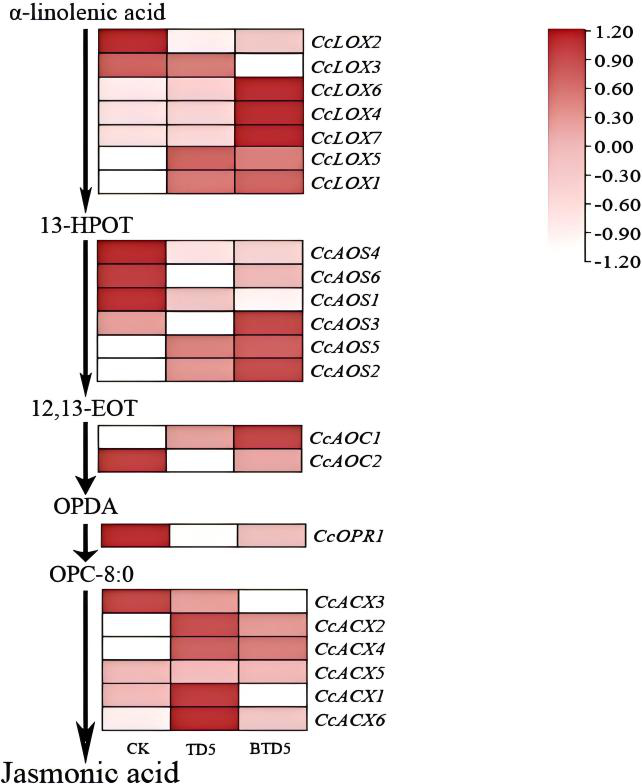
. Expression profiling of key genes following harvesting
The analysis of gene expression levels (Figure 13) illustrated the dynamic changes in genes involved in the JA biosynthetic pathway. On the fifth day post-harvesting (BT-D5), the expression of CcLOX4-5, CcAOS2-3, CcAOS5, CcAOC1, CcACX1-2, and CcACX4-6 increased significantly compared to the CK group. In the wound-dressing treatment (T-D5), although the expression levels of CcLOX4-5, CcLOX7, CcAOS2-3, CcAOS5, CcAOC1, and CcOPR1 were higher than those in the CK group, they were generally lower than those observed in the BT-D5 group. Furthermore, no significant differences were detected in the expression of CcACX2, CcACX4, and CcACX5 between the T-D5 and BT-D5 groups. CcLOX6 and CcAOS6 maintained stable low-level expression across all treatment groups, whereas the expression of CcLOX2-3 and CcAOC2 exhibited a downward trend following harvesting.
. Discussion
Mechanical wounding induced by harvesting is a critical stress factor that limits growth and resource accumulation in Cinnamomum species. This study demonstrates that in response to harvesting stress, a substantial number of transcription factors (TFs), including MYB, AP2-EREBP, NAC, and WRKY, are significantly activated in C. camphora. These findings are consistent with the transcriptome profiling of C. kanehirae and other plants under stress, which revealed a common suite of stress-responsive genes and regulatory mechanisms (Du et al., 2022; Muthuramalingam et al., 2018; Taylor, 2023; Wang et al.,2024; Zhang et al., 2022). These findings are corroborated by the integrated metabolomic and transcriptomic analysis of C. cassia, which revealed that similar TF-mediated regulatory networks are essential for orchestrating secondary metabolite variations (Gao et al., 2023). Recent studies have highlighted the pivotal role of MAPK signaling cascades and hormonal cross-talk (e.g., JA and IAA) in orchestrating the physiological recovery of Lauraceae species following injury (Yao et al., 2024; Zhang et al., 2025). These findings suggest that Cinnamomum species possess a conserved defense system in response to stress.
Regarding hormonal regulation, the levels of jasmonic acid (JA) in the BT group were significantly higher than those in the T group (Figure 3). Although JA is traditionally regarded as a signaling molecule that activates defense responses, elevated JA levels are frequently associated with severe stress responses and growth inhibition. Consequently, the high concentration of JA in the BT group more likely reflects a state of physiological stress rather than a direct promotion of the healing process. In contrast, the application of wound dressing effectively reduced the intensity of wound perception by establishing a physical barrier, allowing the T group to maintain moderate JA levels. This “stress-relief effect” mitigated the growth-defense trade-off (Figueroa-Macías et al., 2021; Monson et al., 2022; Ng et al., 2018), thereby enabling the plant to allocate more resources toward the repair process at the site of injury (Li et al., 2018).
Beyond hormonal regulation, a robust antioxidant system is essential for facilitating effective wound repair. Our data revealed that POD activity in the BT group underwent a continuous decline, potentially leading to an unchecked accumulation of reactive oxygen species (ROS) and subsequent oxidative damage at the injury site (Figure 2b). This correlation between declining peroxidase capacity and impaired tissue recovery aligns with observations in other plant morphogenetic systems (Gupta & Datta, 2003). In stark contrast, the T group, protected by the wound dressing, exhibited a distinct recovery in POD activity during the later stages of healing, thereby maintaining cellular redox homeostasis. This physiological stability creates a complementary causal chain which directly supports the superior regenerative capacity observed in the T group.
Transcriptomic data further corroborated the aforementioned physiological logic. Overall transcriptomic activity in the T-D5 group was higher than that in the BT-D5 group; notably, the GO enrichment analysis revealed that differentially expressed genes (DEGs) were significantly enriched in pathways associated with cell wall biosynthesis and membrane remodeling. This indicates that the wound dressing not only serves as a physical barrier but also functions at the molecular level to accelerate structural reconstruction of damaged tissues by upregulating key responsive genes. Such molecular orchestration is consistent with the framework proposed by Ikeuchi (Ikeuchi et al., 2020). The KEGG enrichment analysis further demonstrated that plant hormone signal transduction and the MAPK signaling pathway are the core pathways in response to harvesting. The broader hormone signaling network identified here likely encompasses not only JA but also abscisic acid (ABA) and indole-3-acetic acid (IAA), as suggested by the enrichment of their respective precursor pathways such as carotenoid and tryptophan metabolism (Figure 9). While our results show a synchronous enrichment of JA-related genes and the MAPK cascade, the direct regulatory hierarchy between these specific signals remains to be fully elucidated. Given that this study did not employ hormone-transcriptome co-expression analysis or protein-protein interaction assays, the precise crosstalk between JA and the MAPK switch should be interpreted with caution. These complex signaling interdependencies represent a vital direction for our future research, aiming to decouple the specific roles of various hormones in orchestrating the MAPK-mediated stress response. This aligns with the findings of Zhang (Zhang et al., 2018), who highlighted that the MAPK cascade serves as a critical molecular switch for sensing wound signals and reshapes defense expression by activating downstream transcription factors.
Notably, key genes within the jasmonic acid (JA) biosynthetic pathway exhibited clear transcriptional activity in response to harvesting stress; however, the magnitude of this expression was more moderate in the T-D5 group compared to the heightened response observed in the BT-D5 group. The phylogenetic analysis showed that CcAOS2 and CcOPR1 clustered closely with the core Arabidopsis genes AtAOS and AtOPR3, respectively. This evolutionary conservation suggests that these genes serve as fundamental regulatory components for JA biosynthesis in C. camphora. The activation of these upstream genes likely initiates a downstream signaling cascade, consistent with established JA-responsive mechanisms involving WRKY transcription factors that coordinate stress adaptation across Catharanthus roseus and Arabidopsis thaliana (Schluttenhofer et al., 2014).
. Conclusions
This study integrated physiological and transcriptomic approaches to elucidate the molecular regulatory mechanisms underlying the root sprouting and regeneration of Cinnamomum camphora under harvest-induced wounding management. Our results demonstrate that mechanical damage from harvesting triggers a stress-defense response in the root system, accompanied by the activation of the jasmonic acid (JA) biosynthetic pathway. The application of a wound dressing effectively mitigated the perceived stress intensity by establishing a physical barrier, thereby maintaining JA levels within a moderate range and optimizing resource allocation between defense and growth. Combined physiological and molecular evidence confirms that the wound dressing maintains high antioxidant enzyme homeostasis and activates cell wall remodeling and MAPK signaling cascades. Furthermore, CcAOS2 and CcOPR1 were identified as core regulatory genes in the JA biosynthetic pathway, providing a critical physiological safeguard for wound site repair. The synergy between physiological and molecular regulation enhances the post-harvest adaptability of C. camphora and improves sprouting efficiency, offering a theoretical foundation for the sustainable utilization and silvicultural management of linalool-type C. camphora resources.
