. Introduction
Genetic diversity is the ultimate source of biological diversity. It plays a crucial role in the survival and resilience of various ecosystems, especially in the face of global climate change (Booy et al., 2008). The increasing threat of climate change is particularly dangerous for long-lived organisms, such as trees. Trees’ adaptability and long-term evolutionary potential greatly depend on their genetic resources (Petit & Hampe, 2006). Therefore, genetic aspects should be considered while managing existing forests, especially in the selection and production of forest reproductive material (Ivetić et al., 2016).
In Poland, 76.9% of forests are managed by the State Forests National Forest Holding (Statistics Poland, 2023b), which makes it easier to successfully implement decisions regarding the management of forest genetic resources (FGR). Currently, the protection of FGR is carried out through the implementation of “The program of conserving forest genetic resources and breeding of trees in Poland for the years 2011–2035”. However, FGR can also be protected indirectly in places of broadly understood nature protection, such as national parks. These constitute just a small fraction of the total country area (1%, i.e., 315,234 ha; Statistics Poland, 2023a). Nevertheless, national parks comprise remnants of natural primeval forests that have emerged through centuries of natural selection, making their genetic resources exceptionally valuable.
Black poplar is a wind-pollinated tree native to Eurasian riparian habitats. This dioecious pioneer species, known for its adaptability and rapid growth, is of great ecological and economic importance (Koskela et al., 2004; Šiler et al., 2014). Unfortunately, black poplar faces the threat of extinction in many European countries due to the destruction of riverine forests (Lefèvre et al., 1998). Indigenous black poplar communities have been transformed into hardwood stands dominated by oak, maple, and ash, or replaced by agriculture and plantations of fast-growing poplar cultivars such as Euramerican hybrids (Populus × canadensis Moench) (Lefèvre et al., 2001). Currently, it seems that the biggest problem related to maintaining the black poplar’s gene pool is the lack of natural regeneration. Seeds of black poplar depend on periodic flooding because they are short-lived and require specific soil conditions for germination and further growth (wet, sandy, and sunny areas without competing vegetation) (Guilloy-Froget et al., 2002). For this reason, river regulation has led to the lack of places for the sexual reproduction of this species. Illegal burning of riverside meadows and the growing population of European beaver have further contributed to significant declines in black poplar population size. Fortunately, genetic studies showed that despite the fragmentation of riparian ecosystems, the remaining black poplar trees maintain high levels of genetic variation, preserving their evolutionary potential (Cottrell et al., 2018; Pospíšková & Šálková, 2006; Smulders et al., 2008b; Wójkiewicz et al., 2019, 2021). However, due to the advanced age of many populations, their deteriorating health condition, and the lack of natural regeneration, it is urgent to develop management strategies and programs to protect the genetic resources of black poplar.
Although the preservation and restoration of genetic resources are one of the objectives of the Wielkopolska National Park (WNP) Protection Plan (WNP Protection Plan, 2013a), so far, they have been poorly achieved. Moreover, the WNP Protection Plan only generally refers to the conservation of riparian habitats. However, as stated in the “Ordinance of the Minister of Climate and Environment of January 21, 2022, on protection tasks for the Wielkopolska National Park for years 2022–2023”, specific actions are about to be taken. Among these, due to the joint efforts of the WNP and the Institute of Dendrology, Polish Academy of Sciences, a black poplar clone archive was established at the WNP. As only 18 trees with different genotypes remain in the park itself (19 genotypes found by Lewandowski et al. (2021), but the biggest tree was blown over in 2022; Figure S1), the gene pool of the archive requires enrichment. Therefore, the main goal of this study was to select appropriate black poplars from the vicinity of the WNP to propagate and plant within the archive. For this purpose, we used a set of 18 nuclear microsatellites to genotype black poplar trees from three neighboring groups and verified whether their gene pool differed from the gene pool of black poplar from the WNP. Specifically, we aimed to: (1) identify hybrids and clones to determine the actual number of genetically distinct trees; (2) evaluate whether low population size and dispersion of the studied groups have led to significant genetic differentiation in the studied area; (3) select the best group(s) that could enrich the gene pool of rare alleles of trees from the WNP.
. Materials and methods
. Study area
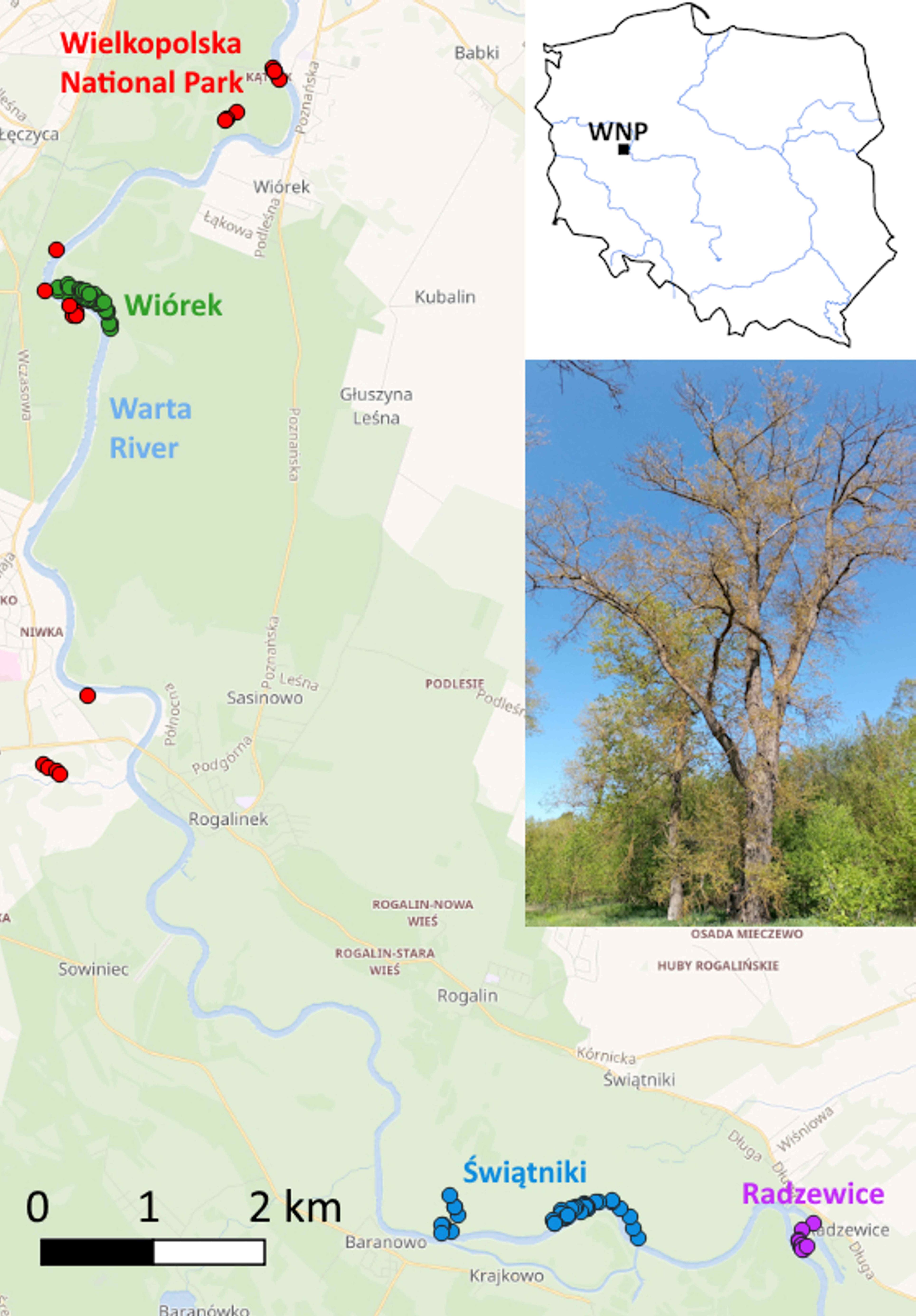
The WNP is one of 23 national parks in Poland. It was established in 1957. The WNP is located in western Poland, which is a part of the Central European Lowlands. The eastern boundary of the WNP runs along the middle section of the Warta River – the third longest river in Poland. The WNP covers 7,597 ha (15,003 ha, including the buffer zone). Isolated black poplar trees remain in the WNP only within the better-preserved sections of riparian forests, adjacent to the Warta River along a roughly 7 km stretch. According to the 2013 report, they occupy only 0.38 ha and are in the VI age class (WNP Protection Plan, 2013a), so at present, they already fall into the VII age class (121–130 years old).
Our study began with a search of the right bank of the Warta River in the vicinity of the WNP to locate groups of black poplars that could be used to enrich the WNP’s gene pool. The entire area we combed through is protected under the Habitats Directive as part of the Natura 2000 network (the Rogalin Valley of the Warta River; habitat code: PLH300012). As a result, we selected three locations: Wiórek (W), Świątniki (S), and Radzewice (R) (Figure 1). We collected fresh leaves from a total of 128 adult black poplar trees. Leaves were stored at −20 °C until DNA extraction. We also incorporated 19 black poplar individuals from the WNP found by Lewandowski et al. (2021). To identify possible hybrid individuals that we could not distinguish by phenotype, we genotyped a set of 19 known reference representatives of Euramerican hybrids and Populus deltoides (W. Bartram ex Marshall).
. Molecular analyses
DNA extraction was carried out using the CTAB protocol (Dumolin et al., 1995), with slight modifications to eliminate RNA contamination. We used approximately 100 mg of frozen material, obtained by grinding the leaf tissue in liquid nitrogen. To remove RNA, an additional step was performed by adding 5 µl of RNase A (10 mg/mL) during the final incubation at 60 °C for 30 min. The quality of extracts was checked based on the results of their electrophoretic separation in a 1% agarose gel. DNA concentration of each sample was adjusted to 15–20 ng/µl using a BioPhotometer plus spectrophotometer (Eppendorf AG, Germany).
We genotyped all samples with a set of 18 nuclear microsatellite markers developed by van der Schoot et al. (2000) and Smulders et al. (2001). Specifically, we used WPMS: 01, 03–10, 12–18, 20, and 22. The markers were amplified in three multiplex PCR reactions according to the protocols described by Wójkiewicz et al. (2019). The amplification products were separated using an ABI3130xl capillary sequencer with GeneScanTM 500 LIZTM internal size standard (Thermo Fisher Scientific, USA). Raw data were scored with GeneMapper ver. 4.0 (Thermo Fisher Scientific, USA) and verified manually. Allele sizes were converted into discrete size classes using Tandem (Matschiner & Salzburger, 2009) and adjusted manually. A set of known genotypes was used as a reference to account for allelic drift between particular runs. The hybrid screening was carried out with win3 nuclear DNA and trnS-trnfM chloroplast DNA markers as described by Wójkiewicz et al. (2019). Win3 displays length polymorphism between P. nigra and P. deltoides that can be detected using a standard PCR. Amplification products, in the case of hybrids, are a combination of patterns characteristic of pure species with extra heavier bands caused by heteroduplex DNA molecules (Heinze, 1997). Win3 is reliable only for assessing first-generation hybrids (Heinze, 1997). TrnS-trnfM is a chloroplast PCR-RFLP marker that enables the detection of hybrids from the second or subsequent generations originating from the maternal P. deltoides lineage (Heinze, 1998). As some microsatellites have alleles characteristic for hybrids (Jelić et al., 2015; Smulders et al., 2008a; Wójkiewicz et al., 2019), the introgression signals were also verified with microsatellite data using STRUCTURE ver. 2.3.4 (Falush et al., 2003; Pritchard et al., 2000) as in Section 2.4 (K = 1–10). We subsequently identified clones using GenClone ver. 2.0 (Arnaud-Haond & Belkhir, 2007). Hybrid and duplicated genotypes were excluded from further analyses.
. Genetic variation and spatial genetic structure
Before we calculated genetic variation parameters, we first estimated the frequency of null alleles in all loci. To this end, we used the maximum likelihood method proposed by Dempster et al. (1977), implemented in FreeNA (Chapuis & Estoup, 2007). The frequency of null alleles was low, ranging from 0.000 in WPMS09 to 0.087 in WPMS22. Hence, the cut-off of 0.190 (Chapuis et al., 2008), above which the values of expected heterozygosity are significantly underestimated, was not exceeded in any case. Therefore, all loci were used in further analyses. Genetic variation parameters were computed for each group using GenAlEx ver. 6.5 (Peakall & Smouse, 2006, 2012). The parameters included the mean number of alleles (A), mean effective number of alleles (AE), number of private alleles (AP), as well as observed (HO) and expected heterozygosity (HE). We used FSTAT ver. 2.9.4. (Goudet, 2003) to compute the inbreeding coefficients (FIS) and mean rarefied allelic richness (AR). Given the presence of null alleles, we subsequently adjusted the FIS values with a Bayesian approach implemented in INEst ver. 2.2 (Chybicki & Burczyk, 2009). The parameters in INEst were set as follows: 200,000 Monte Carlo cycles, updates performed every 20th cycle, and a burn-in of 20,000. The deviance information criterion (DIC) was used to compare the full model (‘nfb’), considering null alleles, FIS > 0, and genotyping failures, with the random mating model (‘nb’), accounting for null alleles and genotyping failures while assuming FIS = 0. This comparison allowed us to test the significance of inbreeding.
To examine the spatial autocorrelation within groups, we used SPAGeDi ver. 1.5 (Hardy & Vekemans, 2002). The average Loiselle’s kinship coefficients (Loiselle et al., 1995) were calculated for pairs of individuals within each group separately. Distance intervals were adjusted to obtain a similar number of pairs in each of the ten distance classes. The statistical significance of the autocorrelations was evaluated through 10,000 permutations. We then constructed the correlograms by plotting the average kinship coefficients against the distance classes. The autocorrelation was significant when the observed data fell outside the 95% CI. To further quantify spatial genetic structure, we calculated the Sp statistics (Vekemans & Hardy, 2004) using matrices of pairwise spatial physical distances and kinship coefficients, generated with SPAGeDi. The relationship between the matrices was evaluated using the Mantel test, implemented in GenAlEx. The significance of the Mantel test was determined with 10,000 permutations.
. Genetic differentiation and clustering
We assessed genetic differentiation by calculating pairwise fixation indexes (FST) and Slatkin’s analogs of FST (RST) in Arlequin ver. 3.5.2.2 (Excoffier & Lischer, 2010). We used the same software to run the analysis of molecular variance (AMOVA). P-values were calculated with 10,000 permutations. To infer the genetic relationships among the studied black poplar groups, we carried out the principal coordinate analysis (PCoA) using GenAlEx. For group clustering, we used STRUCTURE with 500,000 iterations after a burn-in period of 50,000. We selected the admixture ancestry model with correlated allele frequencies, which is suitable for subtle genetic structure (Falush et al., 2003). We set 10 independent runs for K = 1–8. The optimal value of K was determined using the Evanno method (Evanno et al., 2005). Multiple runs for the optimal K value were aligned using the LargeKGreedy algorithm implemented in StructureSelector web-based software (Li & Liu, 2018). StructureSelector was also used to generate STRUCTURE bar plots.
. Demographic history
The possibility of past bottlenecks was tested with two approaches, both available in INEst. First, we used the test developed by Cornuet and Luikart (1996) which assumes that a recent bottleneck leads to the excess of heterozygotes when compared to a population with constant size. The test was run with the parameters recommended by Peery et al. (2012) with 10,000 coalescent simulations. We chose a two-phase model (TPM) that incorporates both single-step and multistep mutations (Di Rienzo et al., 1994). INEst was also used to carry out the test for the deficiency in M-Ratio (MR) (Garza & Williamson, 2001). This test indicates bottlenecks that were more severe and took place in the distant past (Williamson-Natesan, 2005). The simulation of a demographically stable population (MReq) was calculated as the mean of 10,000 coalescence replicates. For both bottleneck tests, we applied the Wilcoxon signed-rank test to calculate p-values with 1,000,000 permutations. In the next step, we estimated effective population sizes (Ne) with the LD approach (Waples & England, 2011), implemented in NeEstimator ver. 2.01 (Do et al., 2014). We set the allele frequency cutoff (Pcrit) to 0.05. 95% CIs were calculated using the parametric option with χ 2 approximation.
. Results
. Genetic variation and spatial genetic structure
We did not detect any cryptic hybrids (Figure 2A). However, we found clones in each group. Clonal genotypes were repeated one to six times and grew nearby (results not shown). Among the studied groups, only W was characterized by a low degree of clonality (genotypic richness, R = 0.90), with moderate values in groups WNP and S (R = 0.68 and R = 0.73, respectively). The highest percentage of clones was detected in group R (R = 0.48) (Table 1).
Figure 2
Bayesian clustering of the black poplar groups analyzed in the study. (A) Bar plot showing results for hybrid detection; (B) bar plot showing results for K = 2; (C) bar plot showing results for K = 3.
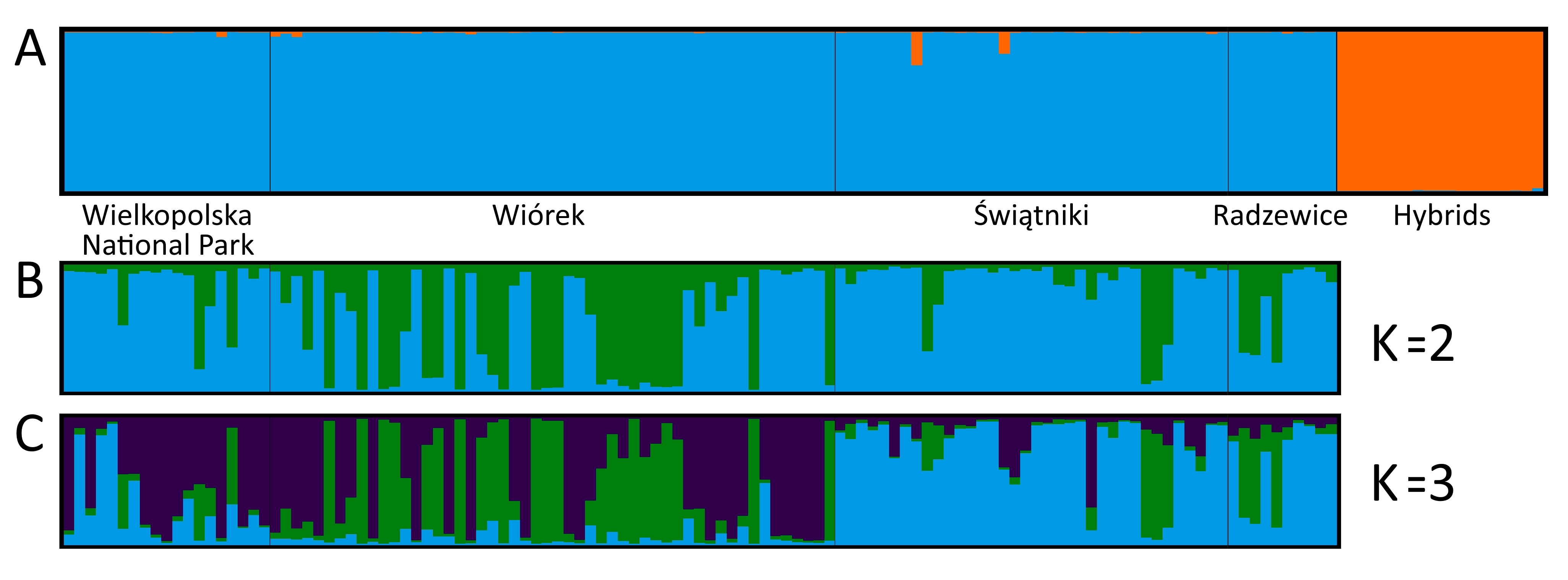
Table 1
Values of genetic variation parameters in the studied groups.
[i] WNP – Wielkopolska National Park; W – Wiórek; S – Świątniki; R – Radzewice; N – number of individuals; NL – number of clonal lineages; R – genotypic richness; A – mean number of alleles; AR – mean rarefied allelic richness; AE – mean effective number of alleles; AP – number of private alleles; HO – mean observed heterozygosity; HE – mean expected heterozygosity; FISnull – inbreeding coefficient corrected for the presence of null alleles; ns – inbreeding is not a significant part of the model; MR – observed M-Ratio; MReq – M-Ratio under mutation-drift equilibrium; * – significantly lower than MReq (p < 0.001); Ne – effective population size.
Groups W and S had a higher mean number of alleles (A = 8.722 and A = 8.833, respectively) and allelic richness (AR = 6.125 and AR = 6.083, respectively) than WNP (A = 7.389; AR = 5.964). The values of the effective number of alleles and private alleles calculated for W and S were close to the values estimated for WNP. In all groups outside the WNP, the observed heterozygosity was higher than expected. Group W was characterized by the highest heterozygosity (HO = 0.763; HE = 0.749). The heterozygosity of WNP and S was only slightly lower. Group R had the lowest values of all parameters. Moreover, the analysis of allele frequency distribution showed that group R differed in this respect from the other groups. In the case of WPMS 03, 13, 16, and 18, the most common allele detected in group R was different than in the other groups (Figure S2). The comparison of full and random mating models revealed that inbreeding was significant in W and R, but its values were near zero (FISnull = 0.012 and FISnull = 0.010, respectively; Table 1). We discovered significant spatial genetic structure in groups WNP, W, and R (Figure S3). A negative correlation was identified in the last distance class in WNP, the eighth distance class in W, and the seventh distance class in R. There was a significant positive correlation in the first distance class in group W. Nevertheless, the values of the Sp statistics were low in all groups (Figure S3).
. Genetic differentiation and grouping
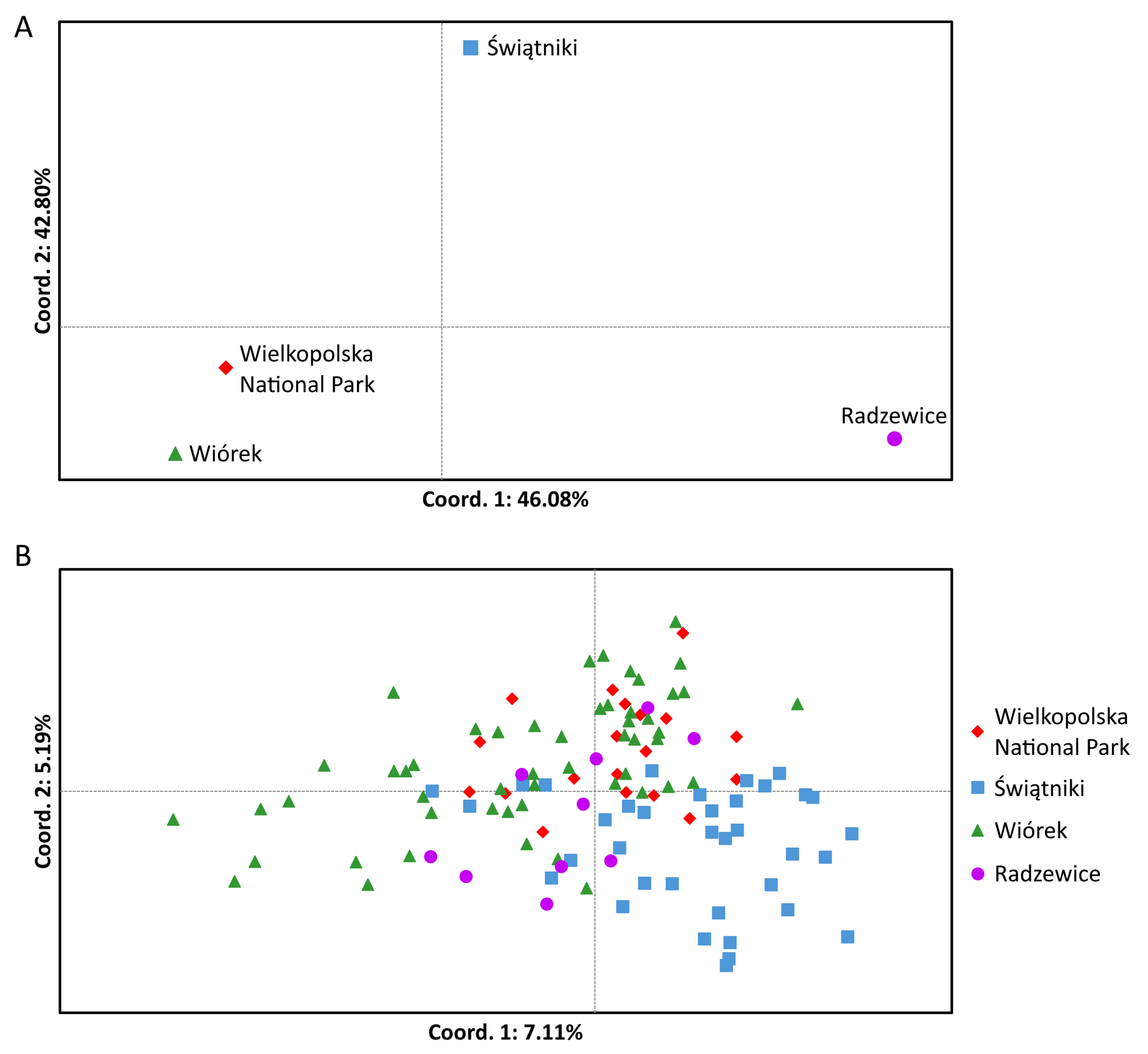
We detected slight genetic differentiation among the studied black poplar groups (FST = 0.0353; RST = 0.0250). Pairwise comparisons revealed significant differences among W, S, and R. The greatest similarity was found between WNP and W, whereas group R was the most different (Table 2). These relationships were better depicted by the results of the PCoA at the group level (Figure 3A). The PCoA carried out at the individual level revealed that in each group outside the WNP, certain individuals were genetically more similar to WNP than the others (Figure 3B). This was also apparent in the results of Bayesian clustering, especially for K = 3. Although the most likely number of clusters was K = 2, K = 3 also had good support (Figure 2B–C).
. Demographic history
We did not detect recent bottlenecks in any group tested. Nevertheless, the calculated MRs were significantly lower than MRseq, indicating that all groups went through a drastic bottleneck in the distant past. Effective population sizes were mostly very low, ranging from Ne = 27.4 for group W to Ne = 109.7 for group S (mean Ne = 64.8).
. Discussion
. Hybrids and clones
Protection of the black poplar’s genetic resources is time-consuming and costly because it requires genetic screening of individuals due to the presence of hybrids and clones. Hybrids are often difficult to distinguish only by phenotypic features and phenology (Bugała, 1973). Nevertheless, molecular analyses confirmed that we managed to successfully exclude hybrids based solely on phenotypic features. However, the actual number of trees that could be used to enrich a clone archive decreased by 30 trees that turned out clones. Poplar genetic clones emerge as a result of damage in different ways: through root suckering, sprouts regrowing from stumps, fallen trees, or broken branches that may be translocated with the river current (Barsoum, 2002; Robak et al., 2024). In our study, clonal individuals were located in close proximity. This indicates that the vegetative spread of black poplar through the Warta’s river current in the vicinity of the WNP is negligible. This is not surprising, because Warta is highly regulated. The very high clonality of group R (R = 0.48) suggests that this stand may have been established artificially. This conclusion was further supported by the lack of spatial genetic structure.
. Genetic variation
Our study showed that black poplar in the vicinity of the WNP retains high genetic diversity. A comparison of allelic variation showed that the mean number of alleles detected in each group depended on the number of trees, as values of rarefied allelic richness were almost the same. On the other hand, there were differences in the effective number of alleles. This means that the studied groups had distinct distributions of allele frequencies. A detailed analysis of allelic patterns revealed differences for group R when compared to WNP, W, and S. This result further supports the non-local origin of group R.
The mean level of heterozygosity found in our study was very similar to the levels estimated for the Polish populations located along the Oder River (Wójkiewicz et al., 2021) and in the middle section of the Vistula River (Lewandowski & Litkowiec, 2017; Wójkiewicz et al., 2019). The same applies to studies conducted in other European countries (Chenault et al., 2011; Čortan et al., 2016; Jelić et al., 2015; Smulders et al., 2008b). Heterozygosity levels in areas of putative glacial refugia of black poplar are only slightly higher (mean HE = 0.820; Pospíšková & Šálková, 2006). Higher heterozygosity was also noted by Çiftçi and Kaya (2019), who analyzed five black poplar populations in Turkey. The authors concluded that this increase in heterozygosity of the Turkish stands was, however, most likely due to a recent bottleneck.
. Demographic history
The values of HO calculated in the groups from the vicinity of the WNP exceeded the values of HE. High levels of heterozygosity, which tend to increase as the trees age, were detected in pine populations (Farris & Mitton, 1984; Yazdani et al., 1985). This observed pattern of heterozygosity is believed to be a consequence of selection due to the fitness advantage of heterozygous genotypes (Ledig, 1986). In our study, tests for the deficiency in M-Ratio were significant in all cases, implying that all groups went through a bottleneck that happened in the distant past. This result is in line with the estimated effective population sizes, which were generally low, especially in groups W and R, for which Ne was lower than 50 (Ne = 27.4 and Ne = 44.3, respectively). The probability of extinction through demographic stochasticity largely depends on the size of a population. Smaller populations have greater chances of becoming extinct due to random events such as natural disasters (Pimm et al., 1988). Moreover, genetic drift diminishes allelic diversity more quickly than in larger populations (Ellstrand & Elam, 1993). According to the 50/500 rule proposed by Franklin (1980), which is now a key indicator in conservation genetics, populations with Ne < 50 are at risk of extinction due to their lower fitness resulting from inbreeding. As a consequence, their genetic variability, survival, and reproductive success decrease with time. To ensure long-term survival, a population should have a minimum Ne = 500. In this way, it can keep a balance between genetic drift and mutations, thus maintaining its evolutionary potential and adaptability (Franklin, 1980). Taking all this into account, even though inbreeding in groups W and R was very small, their very low Ne is alarming.
. Genetic differentiation
The results of AMOVA (FST = 0.0353; RST = 0.0250), as well as pairwise values of FST and RST, point to the slight genetic differentiation among the studied black poplar groups. These values are comparable to the results of previous studies, which, however, covered populations located at greater distances from each other (e.g. Pospíšková & Šálková, 2006). Wójkiewicz et al. (2019) found no significant differentiation between two black poplar populations in the Middle Vistula Valley (Poland), located approximately 100 km apart. Along this section, the river has been only minimally transformed, allowing for free gene flow. Hence, we believe that the differentiation found in our study is above the average for this species and suggests barriers to gene flow in the studied area. The results of the PCoA at the group level indicated that, overall, group W was the most genetically similar to group WNP. This finding was expected, as group W is located next to a group of seven trees from WNP, on the other side of the river.
. Conservation strategy of black poplar in the middle section of the Warta River
Black poplar has little chance of natural regeneration in the transformed ecosystems of the Warta River. This is mainly the long-term consequence of the lake reservoir “Jeziorsko”. Built in 1968, the lake is located at the end of the upper section of Warta and has an area exceeding 42 km2. Its construction limited flooding to a minimum, making the natural regeneration of black poplar virtually impossible (Tylkowski, 2010). It has been a decade since the last greater Warta floods (years 2010 and 2013). Ever since then, the water level has been low (source: Institute of Meteorology and Water Management, Poland).
The Natura 2000 areas in the WNP include alluvial forests (habitat code 91E0). Currently, these habitats are small patches in the valleys of the Warta and Wirynka rivers, with a combined area of 61.90 ha (WNP Protection Plan, 2013b). They are in danger due to degeneration (neophytisation, fruticetisation), inadequate hydrological regimes (such as the absence of flooding), and insufficient amounts of dead wood (WNP Protection Plan, 2013b). Current conservation activities in the 91E0 habitat concern only increasing the dead wood volume and taking phytosociological photographs at 5-year intervals (WNP Protection Plan, 2013b). Furthermore, black poplar in the WNP is directly endangered by the presence of invasive species, such as ash maple (Acer negundo L.). Ash maple exhibits rapid growth, high seed production, drought and frost resistance, as well as substantial phenotypic plasticity, facilitating adaptation to changing habitat conditions. In river valleys, its diaspores disperse with the river current. Compared to other tree species, ash maple also displays relative resistance to flooding and almost immediately colonizes gaps left behind by floods (Chmura et al., 2018). In addition to this, there are many hybrid poplars planted along the roads in the WNP in the middle of the last century, causing the risk of genetic introgression. Individual black poplar trees or their small groups are scattered randomly over the entire area adjacent to the Warta’s riverbed, which further limits gene flow.
Considering the above, we support the decision that the genetic resources of black poplar from the WNP area should be protected ex situ in the form of a clone archive. The archive should consist of at least 50 genetically distinct individuals, with a sex ratio of about 1:1. Group W seems to be the best choice due to its natural character supported by significant spatial genetic structure in the first distant class. It had the highest values of genetic variation parameters but low Ne, so its gene pool also requires action to be preserved. Differences in the gene pools of rare alleles support the inclusion of at least some individuals from group S. This would expand the WNP’s gene pool and thus may increase the adaptive potential of black poplar from the WNP. The results of this study suggest that group R was established by man. We, therefore, recommend not using any individual from group R to enrich the archive.
The WNP’s black poplar clone archive will serve as a source of material for vegetative propagation. It will be used to reintroduce black poplar along the Warta River, both in the park and in the neighboring forest districts (Babki and Konstantynowo). Selected individuals may also be planted in areas where the number and/or sex ratio of the remaining black poplar populations is insufficient for their survival, even though the habitat conditions are still suitable. Such activities fit into the objectives of “The 2030 National Environmental Policy” (PEP2030) in Poland, where great emphasis is put on the protection and renaturation of the most valuable, damaged ecosystems, especially riparian habitats.
. Conclusions
Our work demonstrated the need for genetic testing to select individuals for reproductive purposes, such as to establish a clone archive, even when the chosen objects are in close proximity. This is particularly true in areas affected by human activity and when the range of the species is fragmented. Black poplar not only can reproduce vegetatively but also hybridizes with hybrid poplars, which may lead to the emergence of cryptic hybrids of further generations. Therefore, this species requires routine genetic screening because the actual genetic resources of black poplar are most likely lower than estimated by the number of individuals. Furthermore, our previous paper showed that multi-stem black poplars may have distinct genotypes (Żukowska et al., 2021), which is important for small populations where each genotype counts.
Reestablishment of the declining populations of black poplar is one of the tasks within “The program of conserving forest genetic resources and breeding of trees in Poland for the years 2011–2035”, mentioned in the introduction. As there are no provenance experiments, nor progeny plantations of black poplar in Poland, we will continue our efforts to establish clone archives throughout the country. Without a doubt, the conservation of genetic resources in national parks should be an important element of their long-term protection plans.
. Supplementary material
The following supplementary materials are available for this article:
Figure S1. Photograph of the fallen oldest black poplar tree in the WNP (circumference at breast height: 498 cm).
Figure S2. Allele frequencies of selected markers in the studied groups.
Figure S3. Correlograms of the spatial genetic structure analysis. Loiselle’s kinship coefficient is plotted for ten discrete distance classes with respective 95% CIs (dotted lines).
