Introduction
Hepatica is traditionally known as a small genus within Anemoninae (Ranunculaceae) that comprises seven to nine species depending on the taxonomic concept applied (Tamura,1993; Weiss et al.,2002). Additionally, the taxonomic rank of the genus is also unequivocal; whereas Hoot et al. (2012) subsumed Hepatica to the genus Anemone, Jiang et al. (2017) showed that Anemone s. l. (in this very broad taxonomic sense) is paraphyletic and separated Hepatica with the inclusion of the sections Anemonidium, Keiskea, and Omalocarpus of the genus Anemone s. l., which is also supported by the phylogenetic results of Liu et al. (2018). The partial inclusion of Hepatica and Anemonidium in the dated phylogeny of Pulsatilla (Sramkó, Laczkó, et al.,2019) revealed that their separation took place ca. 21.5 million years ago (Mya). Owing to the morphological evaluation of Hepatica by Tamura (1993), together with the deep split between Hepatica s. s. (in a strict sense) and Omalocarpus + Anemonidium + Keiskea suggested by the latest phylogenetic results, we treated Hepatica in a strict sense (i.e., without the inclusion of related sections as listed above) at the genus level throughout this study.
Two sections are recognized within the genus Hepatica s. s.: the mainly diploid section Hepatica (2n = 14) is characterized by species with entirely lobed leaves, whereas the predominantly polyploid section Angulosa (2n = 28) consists of crenate-leaved species (Zonneveld,2010). The genus shows both inter- and intracontinental disjunct distribution, with the highest species diversity in Northeast Asia (Pfosser et al.,2011). Species of the section Angulosa occur in Central to West China (Hepatica henryi Nakai and H. yamatutai Steward), Central Asia (H. falconeri Thomson), and Europe (H. transsilvanica Fuss). The distribution area of H. transsilvanica is restricted to the Southeastern Carpathians in Romania (Kliment et al.,2016), whereas H. nobilis Mill. – the only species of the genus in Europe that belong to the section with entirely lobed leaves – has a much wider distribution area, which covers most of Europe (Figure S1). Other species of this latter section occur in E Asia and N America (Pfosser et al.,2011, Figure 1).
Polyploid species of section Angulosa were proven to be allopolyploid hybrids of the only diploid but crenate-leaved Central Asian species, H. falconeri Thomson, and other species with entirely lobed leaves (Weiss-Schneeweiss et al.,2007; Zonneveld,2010). The geographic distribution of these species, together with their unique cytotypes and additive genome sizes, led Weiss-Schneeweiss et al. (2007) and Zonneveld (2010) to conclude that the most probable parental species of H. transsilvanica are H. falconeri and H. nobilis. For its conspicuous distribution, H. transsilvanica was the scope of Bartha et al. (2014), who used additive polymorphic sites (APSs) (i.e., unambiguous double peaks in the direct nuclear sequences, where the single-nucleotide polymorphisms (SNPs) of both parent species occur at the same position in the hybrid specimens) of the nuclear region At103. Their results, based on APSs in this genic region, corroborated the findings of previous authors regarding the putative parental species of H. transsilvanica.
From a phylogeographic point of view, the divergence date of H. transsilvanica might have an important biogeographic implication; however, this has never been addressed. The Carpathian Mountains are important sources of genetic diversity for certain species (e.g., Mosolygó-Lukács et al.,2016; Mráz & Ronikier,2016; Slovák et al.,2012). The long-term survival of a broad-leaved forest species in the Carpathians was tested by Bartha et al. (2015), who used Erythronium dens-canis L. as a model species to prove the persistence of a distinct lineage within the species in the Eastern Carpathians. Similarly, Ronikier et al. (2008) discovered a long-term isolation effect within the range of Campanula alpina Jacq., which shaped the genetic variation in the Eastern and Southern Carpathian populations. Moreover, such large disjunctions in species’ distribution, which can be observed in Hepatica section Angulosa, could be associated with deep splits in the phylogeny (e.g., Yokoyama et al.,2000). Lendvay et al. (2016) showed that Syringa josikae J. Jacq., a unique species of lilac native to the Carpathians, can be validated as a Tertiary relict in the European flora. This species survived the eradication of the European Tertiary flora in the Carpathians (in the Ukrainian Carpathians and the Apuseni Mountains in Romania) (Lendvay et al.,2016) and diverged from its closest relatives ca. 1.88 Mya. The distribution patterns displayed by S. josikae and its closest relatives are strikingly similar to the one we can see in Asian tetraploid species of Hepatica section Angulosa.
All previous results hint at the long-term survival of H. transsilvanica in Europe. In our study, we investigated the origin of H. transsilvanica using a molecular phylogenetic approach by including the analysis of two nuclear and two plastid loci. We evaluated the phylogenetic relationship between the species and carried out a divergence date estimation to test the possible autochthon long-term survival of H. transsilvanica in a temporal framework. If the phylogenetic relationships reconstructed using individual gene trees mirrored an allopolyploidization event, we expect discordant topologies that could reveal the parental species of H. transsilvanica (Wendel & Doyle,1998). In the case of autopolyploid origin of H. transsilvanica – as alleles in the polyploid would originate from the same species – we expected gene trees to place this species concordantly in the same phylogenetic position, as sister to its closest extant relative. In both cases, the divergence age of H. transsilvanica could be reconstructed. A relatively old divergence of this species could possibly highlight the role of the Carpathian Mountains in the long-term preservation of this species.
Material and Methods
Sampling
Our field sampling focused on the distribution areas of H. transsilvanica and both of its most probable parental species (Bartha et al.,2014; Weiss-Schneeweiss et al.,2007), H. nobilis and H. falconeri (Table 1, Figure S1). We tried to include samples from different parts of the distribution areas of our focal species to represent the variability within species as much as we could (Figure S1). For H. nobilis, we included samples from the southern part of its distribution area, which was not covered with ice nor too cold for the species to survive during the Last Glacial Maximum (LGM). If this species survived glaciations in Southern European refugia (e.g., Hewitt,1999; Taberlet et al.,1998), sampling along an eastern–western gradient should catch a significant amount of the species’ variability, even with a limited sample size (Table 1, Figure S1). We analyzed one individual from each population, except for H. falconeri, from which species we included two samples from the same population in the analyses of nuclear DNA regions. Samples were dried in silica-gel and stored at room temperature (15–25 °C) prior to DNA extraction. To represent the Asian taxa in some of the phylogenetic analyses, the nuclear ribosomal internal transcribed spacer (nrITS) sequences of H. asiatica and H. henryi were obtained from GenBank. Newly generated sequences for the study were uploaded to GenBank (Table 1).
Table 1
Geographic location of samples with GenBank accession numbers of sequences generated for the study.
Choice of Genic Regions
The nrITS region is widely used in plant phylogenetics (Baldwin et al.,1995) and is described as a suitable DNA region to investigate hybridization and polyploid speciation (Wendel,2000). When analyzing nrITS sequences, it is important to consider that different copies in the nrITS array are homogenized over time by concerted evolution (Álvarez & Wendel,2003; Bailey et al.,2003).
As concerted evolution might distort the phylogenetic signal and we cannot assume that it is complete (Álvarez & Wendel,2003; Feliner & Rosselló,2007), if we want to reliably reconstruct ancient polyploidization events, nrITS should be supplemented with other – possibly low-copy – nuclear markers that are less susceptible to concerted evolution (Sang,2002). The mutL homologue 1 gene (MLH1), which encodes a DNA mismatch repair protein, is documented to be single-copy in a wide range of angiosperms (Zhang et al.,2012) and was proven to be suitable to reconstruct phylogenetic relationships within the genus Pulsatilla (Sramkó, Laczkó, et al.,2019). Therefore, we selected this nuclear region to provide phylogenetic signal from the nuclear DNA besides the widely used nrITS.
To investigate the maternal lineages, we screened commonly used plastid genic regions for polymorphic sites. These regions were the trnLUAA intron with trnL-trnF intergenic spacer (IGS) and four IGS regions (trnH-psbA, petA-psbJ, psbM-trnD, and accD-psaI). After screening on a subset of samples, we selected the trnL-trnF IGS with the trnLUAA intron and accD-psaI for subsequent analyses. Polymerase chain reaction (PCR) primer information for the DNA regions used is given in Table S1.
Laboratory Protocols
Genomic DNA from desiccated leaves were extracted using the E.Z.N.A Plant DNA DS Mini Kit (Omega Bio-Tek; Norcross, GA, USA) following the manufacturer’s instructions. PCRs were conducted in 25-μL final volume mixtures containing 1× Phusion High Fidelity Hot Start II Reaction Buffer (Thermo Scientific; Carlsbad, CA, USA), 0.2 mM of each dNTP (Thermo Scientific), 25 mM MgCl2, 0.25 mg Bovine Serum Albumin (Invitrogen; Carlsbad, CA, USA), 0.2 μM of forward and reverse primers, 0.03 U of Phusion High Fidelity Hot Start II DNA Polymerase (Thermo Scientific), and 25 ng of DNA template. All PCR thermal cycling regimes were performed as specified in Laczkó et al. (2019), except for that of the MLH1 gene, for which we used the same PCR profile as for the nrITS, but set the annealing temperature to 57 °C. Successfully amplified PCR products were sequenced using a commercial service (Macrogen Inc., South Korea).
Reconstruction of Phylogeny and Divergence Date Estimation
As our target species is a known hybrid, we checked all raw electropherograms by eye, not just for obvious sequencing errors, but also for APSs. We trimmed off both ends at the same position and aligned the sequences using MUSCLE v.3.8.31. (Edgar,2004), then phased ambiguous sites of the nuclear sequences using PHASE v.2.1.1. (Stephens et al.,2001; Stephens & Scheet,2005) with default options, for which the format conversion was performed using the SeqPHASE online tool (Flot,2010). The phased sequences of such samples were included in the subsequent analyses as sequence version “a” and “b” of the same samples. The PHI statistics (pairwise homoplasy index) (Bruen et al.,2006) to test for possible recombination events in the nrITS and MLH1 sequences was conducted in SplitsTree v.4.14.4. (Huson & Bryant,2006).
As the molecular evolution of different genic regions can be totally different, in the case of hybrid speciation, concatenating them would easily lead to ambiguous phylogenetic tree topologies. To avoid such a situation, we preferred to study the phylogenetic pattern of gene trees for our selected genic regions separately, and interpreted the conflict between the gene trees as possible evidence for hybrid speciation (Wendel & Doyle,1998). The exception was the plastid sequences (i.e., trnL-trnF and accD-psaI), which are tightly linked and evolve as a single unit representing the maternal lineage (Mogensen,1996); therefore, we concatenated them into a single “plastid” dataset.
Phylogenetic relationships between our focal species were reconstructed by both maximum likelihood (ML) and Bayesian methods. For the ML analysis, we used the online version of PhyML 3.0 (Guindon et al.,2010) with smart model selection (SMS) (Lefort et al.,2017). To assess branch support, besides the nonparametric Shimodaira–Hasegawa approximate likelihood ratio test (SH-aLRT) and bootstrap (bs), parametric aBayes values were calculated; aBayes exhibits the highest statistical power, whereas nonparametric tests are more conservative but (especially aLRT) may be more robust if not all assumptions are met (Anisimova et al.,2011). Nonparametric bootstrap support values were calculated after 1,000 iterations. We interpreted only those branches that were supported by at least two statistics and considered a branch supported if SH-aLRT or aBayes >0.95, and bs >70%.
We estimated the species’ divergence times using BEAST v.2.6.1. (Bouckaert et al.,2019) for nrITS, MLH1, and the plastid dataset separately. To test the presence of a strict molecular clock, we compared the marginal likelihoods of runs that used the strict clock with those that used the uncorrelated relaxed clock. We assessed marginal likelihoods using nested sampling (NS) to compare the probabilities of the clock models by Bayes factors (BFs) – a method demonstrated to have a high statistical power (Maturana-Russel et al.,2018). NS used 10 particles, a chain length of 100 million, and a subchain length of 10 million. To find the optimal substitution model, we applied the Bayesian model test (Bouckaert & Drummond,2017) simultaneously in each run.
For secondary dating, we placed a calibration point at the root, taking the estimates of Wang et al. (2016) as a normal prior with a mean of 27.6 Mya and a sigma of 1.4. Weiss-Schneeweiss et al. (2007) showed that the speciation events within Hepatica occurred in the Pliocene, ca. 3 Mya, but their phylogeny was not well resolved. Divergence dating used a general clock rate and – as admitted by the authors (Weiss-Schneeweiss et al.,2007) – an external calibration point that should be used with caution (see below). Therefore, we used their estimations with a higher margin of error than described and placed a normal prior on the age of Hepatica with a mean of 3.0 and a sigma of 4.0, resulting in a prior age ranging from the present to 10.4 Mya. The gene trees reconstructed by ML were specified as starting trees, but without any multimonophyletic constraints. BEAST v.2.6.1. (Bouckaert et al.,2019) was run two times on each dataset for 500 million generations, saving every fifty thousandth sample. Plastid regions were allowed to have a different substitution model, but the clock rate and the resulting trees were linked in all analyses. We checked the runs for effective sample size (ESS) and convergence using Tracer v.1.6.0., which were accepted if the ESS was greater than 200 with 10% burn-in. We combined runs with LogCombiner v.2.6.1. (Bouckaert et al.,2019) and checked postburn-in trees with Densitree v.2.2.7. (Bouckaert et al.,2019). We reconstructed the maximum clade credibility (MCC) tree with TreeAnnotator v.2.6.0. (Bouckaert et al.,2019). We interpreted not just the MCC tree, but the 95% highest posterior density (HPD) topologies that were estimated by TreeLogAnalyser v.1.10.4. (Suchard et al.,2018).
Results
Sequence Variation
The aligned nrITS data matrix consisted of 568 positions, 90 of which were variable and 58 informative, including the outgroup. Within the ingroup, we identified 19 variable and 10 informative sites. We found only one double peak in the ITS1 region of “H. transsilvanica 2” (Table S2), which was not an APS, but was successfully phased. The PHI statistics found no evidence for recombination (p = 0.888).
The MLH1 matrix was 318-bp long. Forty-seven sites were variable and 24 informative across the entire data set; within the ingroup, we found nine variable and eight informative SNPs. We found four positions where double peaks could be identified (Table S2), but these were restricted to the sequences of H. transsilvanica. All ambiguous sites were successfully phased, except for one (position 41 in the aligned matrix) that was only found in “H. transsilvanica 3” and coded by the IUPAC nucleotide ambiguity codes in subsequent analyses. We found two positions that showed an additive pattern between H. nobilis and H. falconeri. One of them was a consistent APS within all H. transsilvanica samples, whereas the other appeared to be additive only in “H. transsilvanica 2” and “H. transsilvanica 3.” The PHI statistics showed no evidence for recombination (p = 0.055).
Aligned length of the concatenated accD-psaI, the trnLUAA intron and the trnL-trnF IGS (i.e., the plastid dataset) was 1,275-bp long and contained 33 variable and 30 informative SNPs in total. Within the ingroup, we found eight variable positions, four of which appeared to be informative.
ML Phylogenetic Reconstruction
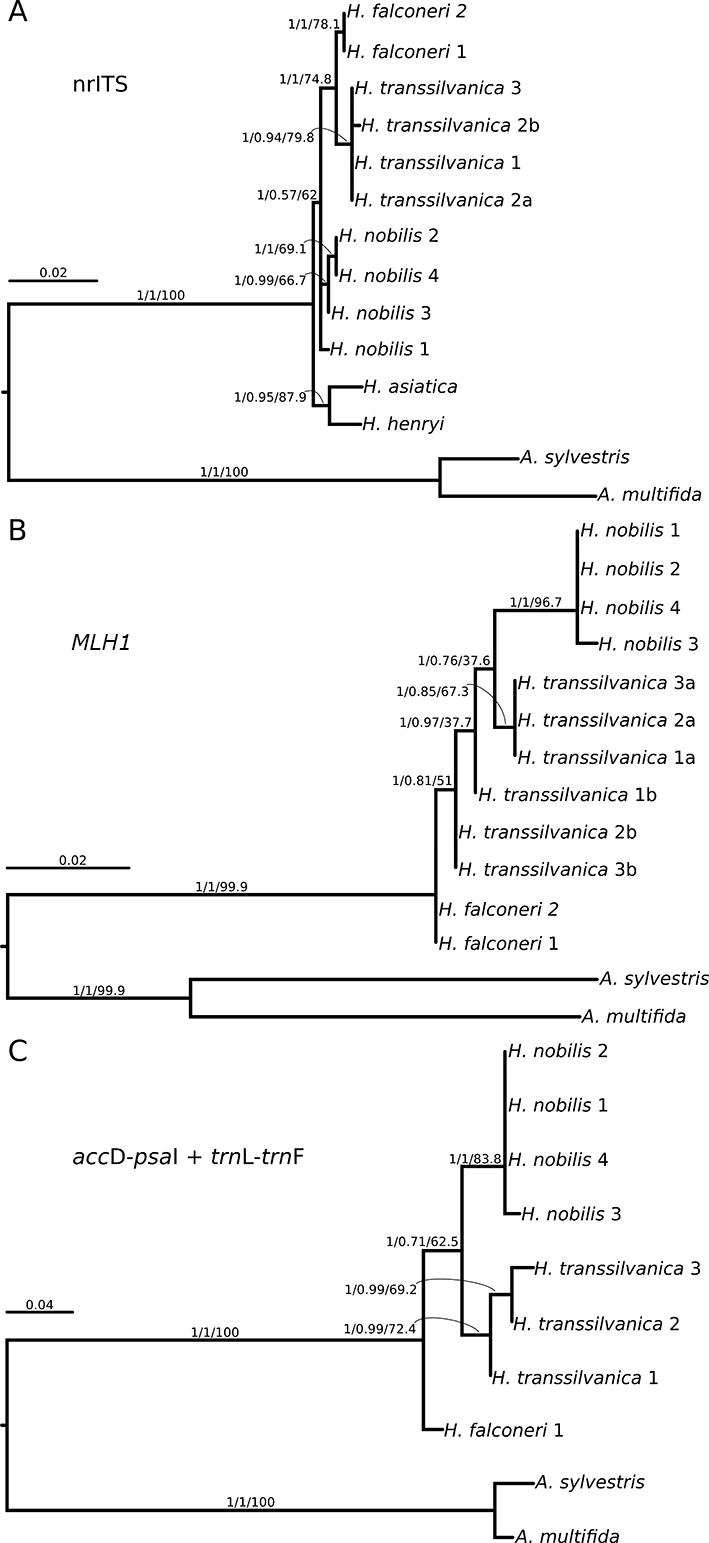
All of the gene trees reconstructed by PhyML separated the outgroup species with high certainty but resulted in different topologies in the placement of H. transsilvanica (Figure 1).
SMS identified the GTR model as the best fitting model of sequence evolution for nrITS. This analysis identified H. asiatica and H. henryi as sister taxa with high support. Samples of H. nobilis were placed on a short branch and formed a metaphyletic group, where the separation was supported only by SH-aLRT. The separation of H. falconeri and H. transsilvanica as sister species received high statistical support (Figure 1A).
For the MLH1 dataset, the HKY85 + G model was identified as the best fitting by SMS. Two sequences of H. transsilvanica (“H. transsilvanica 2b” and “3b”) were grouped together with H. falconeri (Figure 1B). As we move toward the tip of the tree, an intermediate group (“H. transsilvanica 1b” and “H. transsilvanica 1a–3a”) within H. transsilvanica could be identified, although their separation was only supported by SH-aLRT and aBayes (SH-aLRT: 1, aBayes: 0.97). The separation of H. nobilis on a relatively longer branch received high statistical support. In sum, MLH1 clearly placed all phased H. transsilvanica samples as intermediates between its putative parents, although the phylogenetic relationship between these intermediate lineages could not be resolved with statistical confidence (Figure 1B).
The best fitting nucleotide substitution model for the plastid dataset was GTR + G. All branches leading to species received high statistical support. Although H. transsilvanica and H. nobilis are placed as sister taxa, the monophyly of this clade could not be confirmed by statistical tests, except for by SH-aLRT (Figure 1C).
Figure 1
Phylogenetic relationships reconstructed by PhyML using the smart model selection (SMS) for (A) nrITS, (B) MLH1, and (C) concatenated plastid sequences. Statistical support values above branches correspond to Shimodaira–Hasegawa approximate likelihood ratio test (SH-aLRT)/aBayes/bootstrap (bs) values.
Estimation of Divergence Dates
We successfully estimated the divergence time of the target species using a Bayesian approach as implemented in BEAST v.2.6.1. (Bouckaert et al.,2019). The advantage of this approach is that it is not only able to return the MCC tree, but the 95% HPD of tree topologies can also be examined.
For the nrITS dataset, NS supported the presence of the strict clock over the uncorrelated relaxed clock model, whereas for the MLH1 sequences, the presence of a strict molecular clock was rejected. Standard deviations of the marginal likelihood estimates for the nrITS and MLH1 sequences were much lower than the differences in marginal likelihood values (Table S3). NS was not able to distinguish between the likelihood of clock models for the plastid dataset, as the standard deviation of likelihood values were approximately half that of the BF comparing the models (Table S3). Given that the 95% HPD of trees and node ages were overlapping for the plastid dataset with different molecular clock settings, we present our results based on the uncorrelated relaxed clock, which had a slightly better marginal likelihood in our NS analysis.
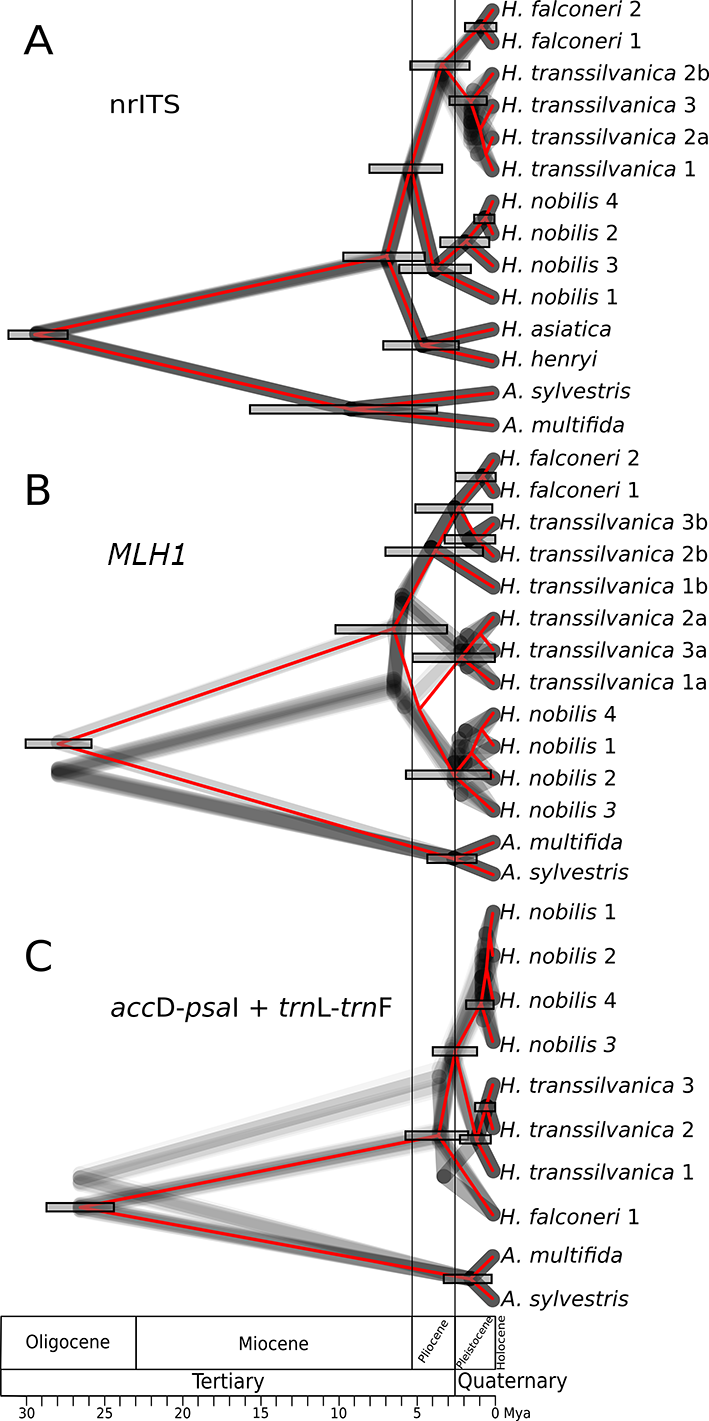
The 95% HPD tree set of the nrITS dataset consisted of 24 unique trees, all of which had the same backbone topology (Figure 2A). The estimated root age was 29.4 million years (My) (95%HPD = 27.5–31.4 Mya). The first branch within Hepatica separated H. asiatica and H. henryi as sister species, which shared a most recent common ancestor (MRCA) with H. nobilis, estimated to lived ca. 6.7 Mya (95%HPD = 4.3–9.7 Mya) in the Messinian age of the Miocene epoch. The next diversification event [5.2 Mya (95%HPD = 4.3–9.7 Mya) in the early Pliocene] separated H. nobilis. This analysis revealed a deep split within H. nobilis, as the MRCA for the samples of this species could be found roughly at the transition of the Zanclean and Piacenzian ages of the Pliocene, 3.8 Mya (95%HPD = 1.4–6.2 Mya). Hepatica transsilvanica was placed as sister species to H. falconeri. The split between these two species is estimated to be 3.2 My old (95%HPD = 3.0–7.8 Mya), which corresponds to the late Pliocene. The 95% HPD trees differed from each other only in their structure within H. transsilvanica, where no equivocal structure could be identified.
The same analysis of the MLH1 sequences contained 15 unique trees and revealed two different backbone topologies (Figure 2B). With an estimated root age of 27.8 My (95%HPD = 25.9–29.8 Mya), the divergence age of Hepatica appeared to be 6.4 My old (95%HPD = 3.1–10.0 Mya). In agreement with the MCC tree, 78% of the trees placed three copies (copies “a”) (Figure 2B) of three different samples of H. transsilvanica as sister to H. nobilis with an MRCA age of 4.7 My (95%HPD = 2.1–8.6 Mya). The remaining trees identified H. nobilis as sister to all the other samples and copies “a” as sister to all other sequences of H. falconeri / H. transsilvanica with an MRCA age of 5.7 My (95%HPD = 2.4–8.6 Mya). The other three copies of H. transsilvanica (copies “b”) (Figure 2B) are grouped together with H. falconeri, although only two of them (“H. transsilvanica 2b” and “H. transsilvanica 3b” from Haţeg and Piatra Craiului, respectively) formed a monophyletic clade. The divergence of “H. transsilvanica 1b” from “H. transsilvanica 2b” / “H. transsilvanica 3b” was estimated to be 3.5 My old (95%HPD = 0.8–6.8 Mya), whereas the MRCA of H. falconeri and “H. transsilvanica 2b” / “H. transsilvanica 3b” can be found at the transition of the Pliocene/Pleistocene 2.4 Mya (95%HPD = 0.2–4.8 Mya).
We found two alternative topologies in the 95% HPD tree set of plastid sequences, which consisted of 24 trees (Figure 2C). The root age of the tree was 26.5 My (95%HPD = 24.5–28.4 Mya). The MCC tree and 63% of the tree set placed H. falconeri as sister to H. nobilis / H. transsilvanica, with an MRCA in the middle Pliocene, 3.5 Mya (95%HPD = 1.8–5.4 Mya). Considering this topology, H. transsilvanica diverged from H. nobilis 2.4 Mya (95%HPD = 1.2–3.7 Mya), roughly at the transition of the Pliocene/Pleistocene, whereas the alternative topology groups H. falconeri and H. transsilvanica together on a short branch with an MRCA of 3.2 My (95%HPD = 1.5–4.9 Mya).
Figure 2
Dated phylogeny of a setof species as reconstructed by BEAST v.2.6.1. 95% highest posterior density (HPD) topology is visualized as grey overlapping phylograms. The maximum clade credibility (MCC) tree is projected on the tree set in red color. Error bars are only given for nodes with a minimum posterior of 0.5 and represent the 95%HPD age of the node. The geographic time scale is placed below the tree in the corresponding places.
Discussion
In this study, we investigated the phylogenetic position and divergence time of H. transsilvanica and its presumed parental species. Our results based on a wider set of genic regions agree with Weiss-Schneeweiss et al. (2007) in the placement of our focal species. Based on nrITS, species occurring in Central and Eastern China (H. asiatica and H. henryi) are placed on a distinct lineage sister to the rest. European taxa (H. nobilis, H. transsilvanica) are assessed to be monophyletic with the Central Asian H. falconeri. Phylogenetic analysis of nrITS placed H. nobilis as sister to H. falconeri / H. transsilvanica. In contrary, plastid data suggest that H. transsilvanica is closest to H. nobilis and H. falconeri is the sister taxa of European species. Although Zonneveld (2010), based on its smallest genome size, hypothesized H. falconeri to be the most probable basal taxon within Hepatica, this assumption is not supported by the placement of H. falconeri on our nrITS tree (Figure 2A), as Eastern Asian species clearly diverged earlier.
The divergence times estimates found in our analyses apparently support the previous finding of Weiss-Schneeweiss et al. (2007) that the Pliocene played an important role in the speciation events of Hepatica, but we found older estimated dates. Previously described crown age (i.e., the MRCA age of a given group of samples) of Hepatica based on nrITS sequences and a wider taxonomic sampling lies between ca. (4.3–)2.5(−0.8) and (0.9–)0.5(−0.2) Mya, but our estimation suggests a 6.7-My-old (95%HPD = 4.23–9.7 Mya) divergence for the ingroup (i.e., Hepatica s. s.), even though we used a narrower set of samples. A similar estimate is achieved using MLH1 sequences but without the inclusion of any East Asian species. The discordance in the timing of isolation events between our study and the previously published analysis might be caused not only by the different genic regions used – as Weiss-Schneeweiss et al. (2007) also used nrITS – but the different strategy and calibration point for divergence dating. Weiss-Schneeweiss et al. (2007) used a general clock rate for the genic regions included in their study and the age of Ullung Island – the distribution range of H. maxima Nakai – as an external calibration point (1.8 Mya), which can be interpreted as both the minimum or maximum age. Moreover, instead of nonparametric rate smoothing (NPRS) and penalized likelihood (PL), we estimated species divergence times using a Bayesian phylogenetic method, as implemented in BEAST v.2.6.1., and applied a secondary calibration point suggested by the fossil calibrated phylogeny of Wang et al. (2016). The estimated divergence times in our study are in accordance with the divergence time of other Eurasian species with similar distribution patterns. The fossil evidence clearly demonstrates that such species were distributed much more widely throughout Eurasia before the Quaternary (e.g., Lendvay et al.,2016; Yokoyama et al.,2000).
These results suggest that a re-evaluation of the evolutionary history of the genus and the possible driving forces of speciation, including hybridization within Hepatica, should be the subject of future studies. Another intriguing result might be the crown age for H. nobilis found in our analyses (mean = 3.8 Mya, 95%HPD = 1.4–6.2 Mya) that correspond to the middle Pliocene. A phylogeographic study using H. nobilis as a model species could potentially reveal important aspects of the phylogeography of the European temperate flora; this broad-leaved forest species is present in Europe from the onset of the Quaternary period. These evolutionary relationships, both at the genus and species levels, could be potentially investigated by the usage of a genomic approach that can provide an in-depth phylogenetic resolution, even if genetic differentiation is shallow (e.g., Sramkó, Paun, et al.,2019; Twyford & Ennos,2012).
Still, our results provide evidence of the origin of H. transsilvanica. The hybrid origin of this species is corroborated by the incongruent phylogenetic placement (Wendel & Doyle,1998) of our samples based on nrITS and plastid sequences, as already postulated by the molecular results of Weiss-Schneeweiss et al. (2007). Using the low-copy nuclear region MLH1, the hybrid speciation event could be reconstructed directly, as the MCC tree placed copies “a” and “b” of H. transsilvanica on two lineages, each affiliated to the lineage of the putative parental species (Figure 2B), H. nobilis and H. falconeri. The paraphyletic placement of copies “b” and the topological incongruence of copies “a” (Figure 2B) might be the result of reticulation, although the PHI statistics did not find significant evidence of recombination (p = 0.055). The statistical uncertainty of placement of copies “a” can be the result of molecular evolutionary processes (e.g., gene conversion) shifting maternal gene copies toward the paternal copies. The overall paraphyly of MLH1 copies (Figure 2B) might be a faithful representation of a 3-My-old gene conversion process in an allopolyploid species. Our analysis of plastid sequences (Figure 2C) supported the hypothesis (Weiss-Schneeweiss et al.,2007) that H. nobilis is the maternal parent of H. transsilvanica. It seems unlikely that another Asian taxon might have played a role in the speciation of H. transsilvanica as the divergence of the hybrid species is estimated to be much younger than the MRCA of H. asiatica and H. nobilis, two species with entirely lobed leaves.
The long-term survival of H. transsilvanica in the Southeastern Carpathians is strongly supported by our results. Dating analyses of three genetic regions independently estimated that the isolation of H. transsilvanica from its paternal parent (i.e., H. falconeri) took place 3.5–3.2 Mya, as assessed from MLH1 “b” copies and nrITS, respectively. The maternally inherited plastid regions revealed the separation of H. transsilvanica from its maternal parent (that is, H. nobilis) took place 2.4 Mya. The significantly lower effective population size of the plastid genome compared to the tetraploid nuclear genome (Schaal et al.,2000), together with the lack of recombination, allow plastid genes to respond more instantly to isolation and give a more accurate date of divergence. Thus, the isolation of the hybrid H. transsilvanica coincides with the start of Quaternary glaciation cycles. Conclusively, all of our analyses using the molecular clock suggested that the MRCA of H. transsilvanica and the parental species existed no later than the early Pleistocene, a period during which a large portion of the Tertiary flora in Europe was reported to have been eradicated (Svenning,2003; Willis & Niklas,2004).
On this background, it is safe to conclude that H. transsilvanica is a relict of the Tertiary Flora that was able to survive the Quaternary climatic changes in the Southeastern Carpathians. The biogeographic importance of this region has been proven before (Mráz & Ronikier,2016); however, the temporal aspect of phylogeography was not always reconstructed (e.g., Bartha et al.,2015; Ronikier et al.,2008). Similarly to the example of Syringa josikae (Lendvay et al.,2016), our results not only highlight the biogeographic role of the Carpathians during the Quaternary glaciations but show the potential of this mountain range in the long-term preservation of Tertiary lineages in Central Europe. Although exact phylogeographic relationships within H. transsilvanica remain to be revealed, its refugium could be associated with calcareous bedrocks. The long-term climatic stability of this mountain range made the survival of endemic species possible in heterogeneous habitat types (see Hurdu et al.,2016 for details). Several broad-leaved forest “dacian” endemic species (i.e., those Fagion species with a distribution centered on Transylvania), such as Aconitum moldavicum Hacq. ex Rchb., Dentaria glandulosa Waldst. & Kit., Symphytum cordatum Willd., and Pulmonaria rubra Schott, etc., can similarly be remnants of a stable glacial refugium in the Eastern Carpathians region (Hendrych,1981).
Handling Editor
Agnieszka Popiela, University of Szczecin, Poland; https://orcid.org/0000-0001-9297-0538
