. Introduction
Membrane-bound organelles such as mitochondria and chloroplasts are present in plant cells and are believed to have originated from endosymbiotic bacteria-like organisms around two billion years ago (Palmer & Delwiche, 1998). One significant evidence supporting this theory is the presence of organellar genomes, which are double-stranded closed circular molecules of DNA that are unique to mitochondria and chloroplasts. In most plants, organellar genomes are inherited uniparentally, meaning that only one parental genome is passed down. This inheritance pattern has allowed us to establish connections between individuals and species. However, there are exceptions to this pattern, as observed in some gymnosperms, including Cryptomeria japonica, Larix decidua and Larix leptolepis, that exhibit possible biparental inheritance, while Biota orientalis, Calocedrus decurrens, Picea pungens, Picea glauca, Pinus contorta × banksiana, Pinus taeda, Pseudotsuga mezniesii and Sequoia sempervirens demonstrate paternal inheritance (Reboud & Zeyl, 1994).
Organellar genomes can differ from each other within a cell or between cells within an individual, resulting in more than one variant of the genome. This phenomenon is called heteroplasmy and was initially detected in plastids and has been observed in almost all clades of the green plant tree of life, as well as in the mitogenomes of both plants and animals (Kmiec et al., 2006; Ramsey & Mandel, 2019). Heteroplasmy can result from several mechanisms, including gene rearrangements, gene chimeras induced by recombination, insertions or deletions, and point mutations. Moreover, in cases of biparental inheritance, in which the parents have different plastomes or mitogenomes, heteroplasmy can also occur in the offspring. Heteroplasmy can manifest visibly, such as in the green and white patterns seen in variegated leaves, where the green pattern results from healthy green plastids and the white pattern arises from diseased or genetically distinct colorless plastids (Chiu et al., 1988).
In the study of the plastid genomes of Phoenix dactylifera cultivars, two mechanisms of plastid heteroplasmy are mentioned (Sabir et al., 2014). The first is biparental inheritance, which means that the offspring inherit organelles from both parents. The second mechanism mentioned is incomplete sorting in uniparental inheritance, which is most likely in date palms due to their maternal inheritance of the plastid genome. Incomplete sorting occurs when plastids are incompletely sorted, and the gametes of the parents are heteroplasmic. There are, of course, other reasons why heteroplasmy occurs in chloroplasts. For example, mutations in some plant species of the genus Medicago presented two types of heteroplasmy, the first resulting from a single base pair change and the second from indels occurring within individuals and between species (Johnson & Palmer, 1989).
The plant mitochondrial genome is more complex than the plastid genome. In particular, the vascular plant mitogenome is larger than that of animals, e.g., the size of the watermelon mitogenome is 2,500 kb, whereas that of animals is between 16 and 20 kb (Palmer & Herbon, 1987; Ward et al., 1981). In addition, the plant mitogenome can vary greatly in structure compared to animals, which have circular mitogenomes and can be present in circular, linear, or complex molecular forms (Kmiec et al., 2006). Heteroplasmy can be caused by independent mutations or biparental inheritance, as in the case of plastid genomes (Wolfe & Randle, 2004), or by mutation, recombination and paternal leakage (Ramsey & Mandel, 2019). Furthermore, it seems that a transfer of the plastid genome to the mitogenome is rather common in vascular plants, which is the next cause of the mitochondrial genome heteroplasmy (Park et al., 2020; Szandar et al., 2022).
To detect heteroplasmy in the liverwort organellar genomes, we apply the latest nanopore sequencing technology, which enables unbiased identification, quantification, and phasing of haplotypes. The presence of heteroplasmy in the liverworts was not studied so far, assuming uniparental inheritance (Pacak & Szweykowska-Kulińska, 2003), but recent discoveries in vascular plant organellar genomics (Lee et al., 2020; Wang & Lanfear, 2019) encourage us to undertake this work. To verify the presence of heteroplasmy in the liverworts we selected four species representing four major lineages, including Riccia fluitans (Marchantiopsida - complex thalloid), Apopellia endiviifolia (Pellidae - simple thalloid I), Aneura pinguis (Metzgeriidae - simple thalloid II) and Scapania undulata (Jungermanniidae - leafy liverworts).
The main aims: sequence, assembly and annotation of organellar genomes using nanopore technology (i), analyzing liverwort organellar genomes towards the presence of structural heteroplasmy (ii), and identification of intraindividual variation within plastomes and mitogenomes (iii).
. Material and methods
. Plant material
Four species of liverworts were collected from Poland with an expedition of Riccia fluitans, which was bought from a commercial company (Table 1). To exclude the possibility of inter-individual variation, collected species were cultured in vitro to obtain the required amount of plant material from a single individual. Based on literature data, three types of media were selected and tested for in vitro cultivation: solid half-strength Gamborg’s B5 medium basal salts (Gamborg et al., 1968) including organics and vitamins – ½GB5 medium (Althoff et al., 2022), ½GB5 medium containing 100 µg/mL cefotaxime – ½GB5+C medium (Althoff & Zachgo, 2020), solid half strength MS medium basal salts (Murashige & Skoog, 1962) including LS medium organics and vitamins (Linsmaier & Skoog, 1965) – ½MS+ovLS medium (Pence et al., 2005). The medium on which the best plant growth was observed was selected for the study, and minor modifications were introduced. Finally, the liverworts grew on the ½GB5 medium with 20 g⋅l−1 sucrose, 8 g⋅l−1 agar–agar and pH 6.0. The upper fragments of sterile plants were used as secondary explants and were placed on the medium in the form of five small clumps separated by approx. 1–2 cm. Plants were grown in climate chambers at 24 °C under long-day conditions with a 16:8 photoperiod (16 h light; 8 h dark).
Table 1
Liverwort species, locality, voucher and organellar genome accession numbers.
. DNA extraction, library preparation and sequencing
Total DNA was extracted using the modified CTAB procedure. Fresh 0.2 g of thallus or leaves was grated in liquid nitrogen, placed in a 12 mL tube, and flooded with 2.5 mL of CTAB1 isolation buffer and 150 µL 2-mercaptoethanol. Next, 5 µL of RNAse was added at conc. 100 mg/mL. The samples were mixed by vortexing and incubated in a water bath at 70 °C with frequent stirring. After 2 h, the mixture was brought to room temperature. Then, 2.5 mL of dichloromethane was added and mixed by inverting the tube. Next, the tubes were centrifuged for 30 min at 8,000 rpm. The supernatant was then transferred to a clean 12 mL tube without disturbing the pellet. 5 mL of CTAB2 (precipitation buffer) was added and mixed gently by inverting the tube. After the precipitate had formed, the tubes were centrifuged for 30 min at 8,000 rpm. The supernatant was pipetted except for about 1 ml of the mixture from the bottom of the tube, which was transferred with any precipitates to a 1.5 mL tube. The tubes were centrifuged for 15 min at 14,000 rpm. If precipitate on the tube wall was present, the supernatant was discarded, and the precipitate was then dissolved in 500 µL of 1M NaCl. Next, 500 µL of isopropanol was added and mixed gently. After centrifugation for 30 min at 14,000 rpm, the supernatant was pipetted off, and the pellet was rinsed twice with 500 µL of 70% ethanol. Next, the tubes were centrifuged for 10 min at 14,000 rpm, and the supernatant was carefully removed. Open tubes were dried in a thermoblock at 37 °C, and finally, the pellet was dissolved in 50 µL of water by incubation at 37 °C for 1 h. The purity of DNA samples was assessed spectrophotometrically using a Cary 60 spectrophotometer (Agilent). DNA quantity was estimated using the Qubit fluorometer and QubitTM dsDNA BR Assay Kit (Invitrogen, Carlsbad, NM, USA). DNA quality was checked by capillary electrophoresis using Tapestation (Agilent) with a Genomic kit. If required, the extracted DNA was additionally cleaned and concentrated using Genomic Purification Kit (New England Biolabs, here after NEB) according to the manufacturer’s protocol. Since total genomic DNA contains only 1–5% of mitochondrial and plastid DNA, to enrich this fraction, a Microbial DNA enrichment kit (NEB) was used for Aneura pinguis, Apopellia endiviifolia and Scapania undulata samples. This method enables PCR-free, bead-based target enrichment, which is crucial for obtaining deep coverage and high-quality assemblies of organellar genomes.
The long-read libraries were constructed using Ligation Sequencing Kit SQK-LSK114 (Oxford Nanopore Technologies, hereafter ONT) and NEBNext®; Companion Module for Oxford Nanopore Technologies®; Ligation Sequencing (NEB). The libraries of Aneura pinguis and Scapania undulata were sequenced using MinION Mk1C sequencer and MinION R10.4.1 flowcell, while libraries of Riccia fluitans and Apopellia endiviifolia were sequenced using Promethion P2 solo device and dedicated R10.4.1 flowcell. To obtain the best quality sequences, the generated raw reads were processed with duplex_tools 0.2.20 (ONT) to generate pairs of duplex reads, enabling over Q30 quality sequences. Reads were basecalled using Dorado 0.1.1 software (ONT) with a super accuracy model (SUP) and stereo-duplex mode.
. Organellar genome assembly and annotation
Basecalled reads were assembled into contigs using Flye 2.91 (Kolmogorov et al., 2019) with – meta flag and minimal overlap set to 2,000 bp using 120 compute cores and up to 0.9 Tb RAM. Assembled contigs were mapped to existing reference genomes, with the exception of Scapania undulata mitogenome, where contigs were mapped onto S. ampliata (NC_052751) using minimap2 (Li, 2018). The mapping process revealed that for all the samples analyzed, organellar genomes were completely assembled during the de novo assembly stage. The draft genomes were circularized and remapped using minimap2 to correct possible errors and calculate mean coverage. Corrected genomes were annotated using data from previously sequenced plastomes and mitogenomes using Geneious Prime 2023. The newly assembled S. undulata mitogenome was drawn using OGDraw v3.1 (Greiner et al., 2019).
. Structural variants and SNP detection
Analysis of the presence of structural variants of plastomes within analyzed species was carried out using the Cp-Hap pipeline (Wang & Lanfear, 2019). For each of the assembled plastomes, a fasta file was prepared, including LSC, SSC, and IR regions. The CP-Hap pipeline was run with −t (threads) set to 120, −x map-ont, and −d (minimum distance of exceeding the first and last conjunctions) set to 1,000. BAM mapping files from the assembly step were used to call SNP using CLAIR3 v.1.0.0 (Zheng et al., 2022) and r1041_e82_400bps_sup_v400 model with following parameters: −t = 120, −p − min_coverage = 50, −enable_long_indel. Since long-read technologies are prone to deletion errors, especially within homopolymeric regions, the annotated variants of this kind were removed from further analyses. The distribution of heteroplasmic SNPs was visualized using Circos (Krzywinski et al., 2009).
. Results
The preliminary sequencing runs of liverwort genomic libraries using MinION 10.4.1 flow cells resulted in 2% and 5% of total Gbp mapped on mitogenome and plastome reference genomes, respectively. The enrichment of total genomic DNA towards organellar genomes increased these values fivefold in the case of mtDNA and 10-fold for cpDNA. Total coverage obtained for studied species varied from 165x in Scapania undulata (mitogenome) to 1,579x in Riccia fluitans (plastome).
. Characterization of the newly assembled organellar genomes
The sequenced and assembled liverwort plastomes and mitogenomes share the same structure, gene, and intron content as previously published. The newly sequenced chloroplast genome of Riccia fluitans spans 121,989 base pairs with a GC ratio of 28.9%. The plastome of Riccia fluitans is 10 bp shorter than other sequenced specimens from Poland (MT023021) and 307 bp longer than the Korean sample (NC_042887), with an identity of 99.957% and 99.632%, respectively. Riccia fluitans mitochondrial genome (OR220799) is 185,615 base pairs in length, which is six bp shorter than the second Polish sample (NC_043906) and 25 bp shorter than the Korean accession (MN927134). The genome comprises 74 genes, including 42 protein-coding genes, three rRNAs, 28 tRNAs, and one pseudogene (nad7). The overall GC content of the genome is 42.4%. The pairwise identity between newly assembled samples and those published previously was 99.961% and 99.972% for MN927134 and NC_04390, respectively.
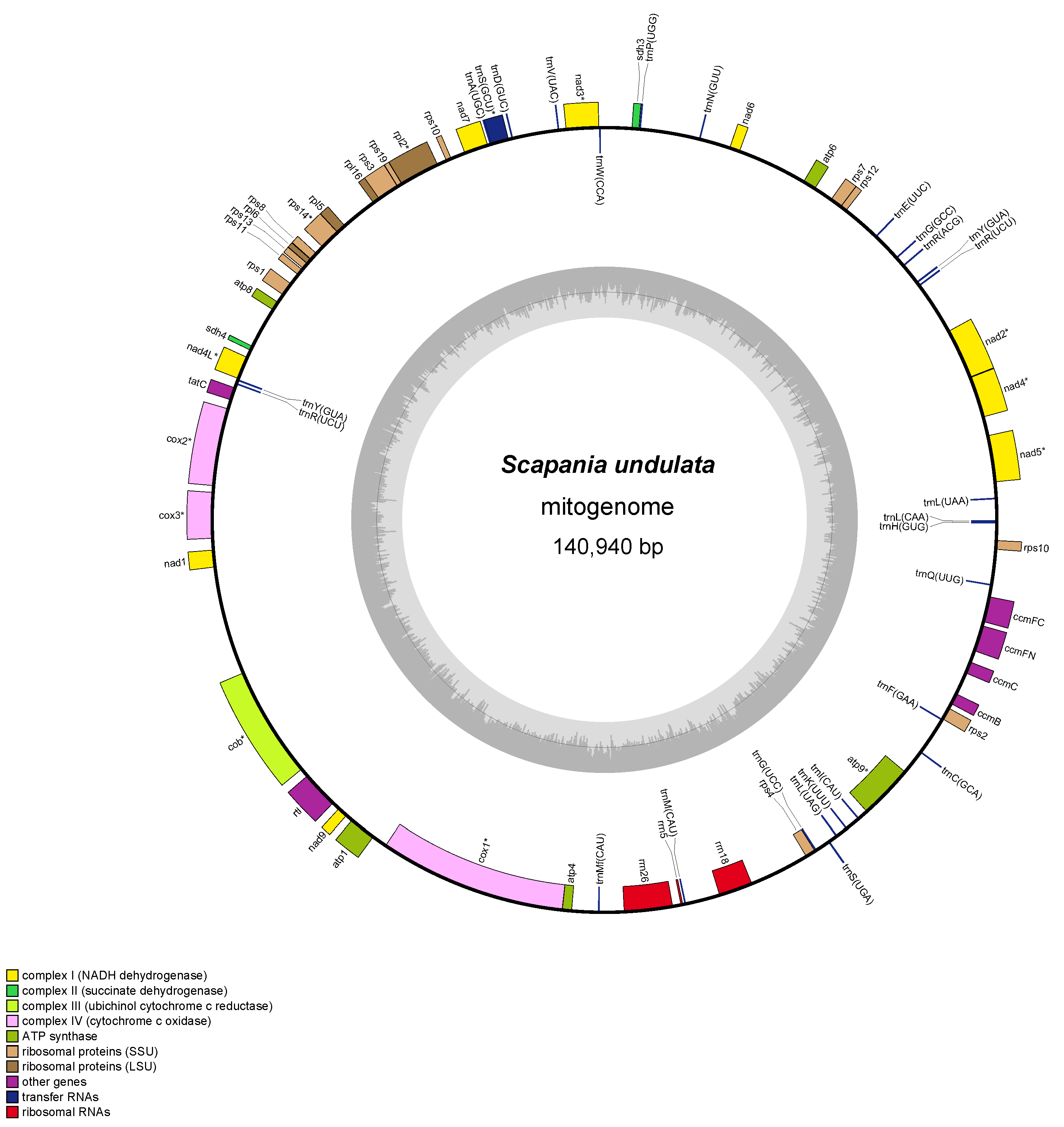
The nanopore-sequenced plastome of Scapania undulata was reported and described in the previous study (Ciborowski et al., 2022). The newly assembled mitochondrial genome is the first report of Scapania undulata mitogenome (GenBank accession number OR220800), which is 140,940 bp long and contains 72 genes (42 protein-coding genes, three rRNAs, and 27 tRNAs) with overall GC content at 45.0% (Figure 1). The gene order of S. undulata mitogenome is the same as that of S. ornithopodioides (MK230950) and S. ampliata (NC_052751), but these mitogenomes are longer by 1,982 bp and 2,724 bp, respectively. The deletions, resulting in the smallest mitogenome within the genus, were not equally distributed but concentrated in three regions: trnY(GUA)-trnR(ACG) - deletion of 753 bp, within nad7 pseudogene - deletion of 1,152 bp and trnV(UAC)-trnD(GUC) - deletion of 417 bp.
Figure 1
Mitochondrial genome of Scapania undulata. Genes inside and outside the outer circle are transcribed in counterclockwise and clockwise directions, respectively. The genes are color-coded based on their function. The inner circle visualizes the G/C content.
The plastid genome of Apopellia endiviifolia sequenced in this study corresponds genetically to the typical form and has the same gene content and order as previously published, with a total length of 120,531 bp, which is the smallest among known plastomes of this cryptic species (Grosche et al., 2012; Paukszto et al., 2023; Sawicki et al., 2021). One hundred twenty-two unique genes (taking into account only one copy of inverted repeat regions) were identified in the plastome of 81 protein-coding genes, four ribosomal RNAs, 31 transfer RNAs, and six ycf genes of an undetermined function. The sequenced A. endiviifolia mitogenome was 109,260 bp long and had the same gene and intron content as previously published mitogenomes of this species. Apopellia mitogenomes are the smallest among the liverworts sequenced so far, but they contain all protein-coding genes and full intron set known from other Jungermanniopsida, including 41 protein-coding genes, three rRNAs, and 26 tRNAs (lost of trnR-UCG).
The analyzed specimen of Aneura pinguis, according to molecular diagnostic characters (Bączkiewicz et al., 2017), belongs to species A and its organellar genomes have identical gene content and order as previously published (Myszczyński et al., 2017). Of the 122 unique genes (i.e., including one copy of the inverted repeats) identified in 120,802 bp long Aneura pinguis plastome, 81 are protein-coding genes, five genes of unknown function (ycf genes), four ribosomal RNAs and 32 tRNAs. The mitogenome of the analyzed A. pinguis sample was 165,319 bp and contained 71 genes, including 40 protein-coding, the rRNAs, and 28 tRNAs.
. Intraindividual variation of liverwort plastomes
Within the plastomes of complex thalloid liverwort, Riccia fluitans, and simple thalloids I, Apopellia endiviifolia, no heteroplasmic SNPs were detected.
Mapping the reads onto the Scapania undulata genome revealed 73 SNP (72 unique, one on the second copy of IR). Most of them were identified within LSC (60), 12 within SSC, and one in the IR region. Among identified SNPs, only four indels were found, including two deletions (two and three bp long) and two insertions (three and five bp long). Both deletions and insertions were located next to each other, deletion in psaI-pafII and insertion in ndhC-trnV(UAC) intergenic spacers. Analysis revealed 69 substitutions, including 29 transversions and 40 transitions. Among the latter, 28 were located in protein-coding genes (CDS), and half of them (14) were non-synonymous. In the case of transversions, all of the 12 located in CDSs were non-synonymous (Figure 2A). Analysis of intraindividual variation of plastome within Aneura pinguis revealed three transitions located in protein-coding genes (Figure 2B). The non-synonymous mutation was found in chlB (G to A) and synonymous in psaB (G to A) and ycf1 (C to T). In the case of both species, detected heteroplasmy was equally distributed along the plastome, with the exception of rps11-trnA(UGC) region, where no infra-individual variation was identified (Figure 3).
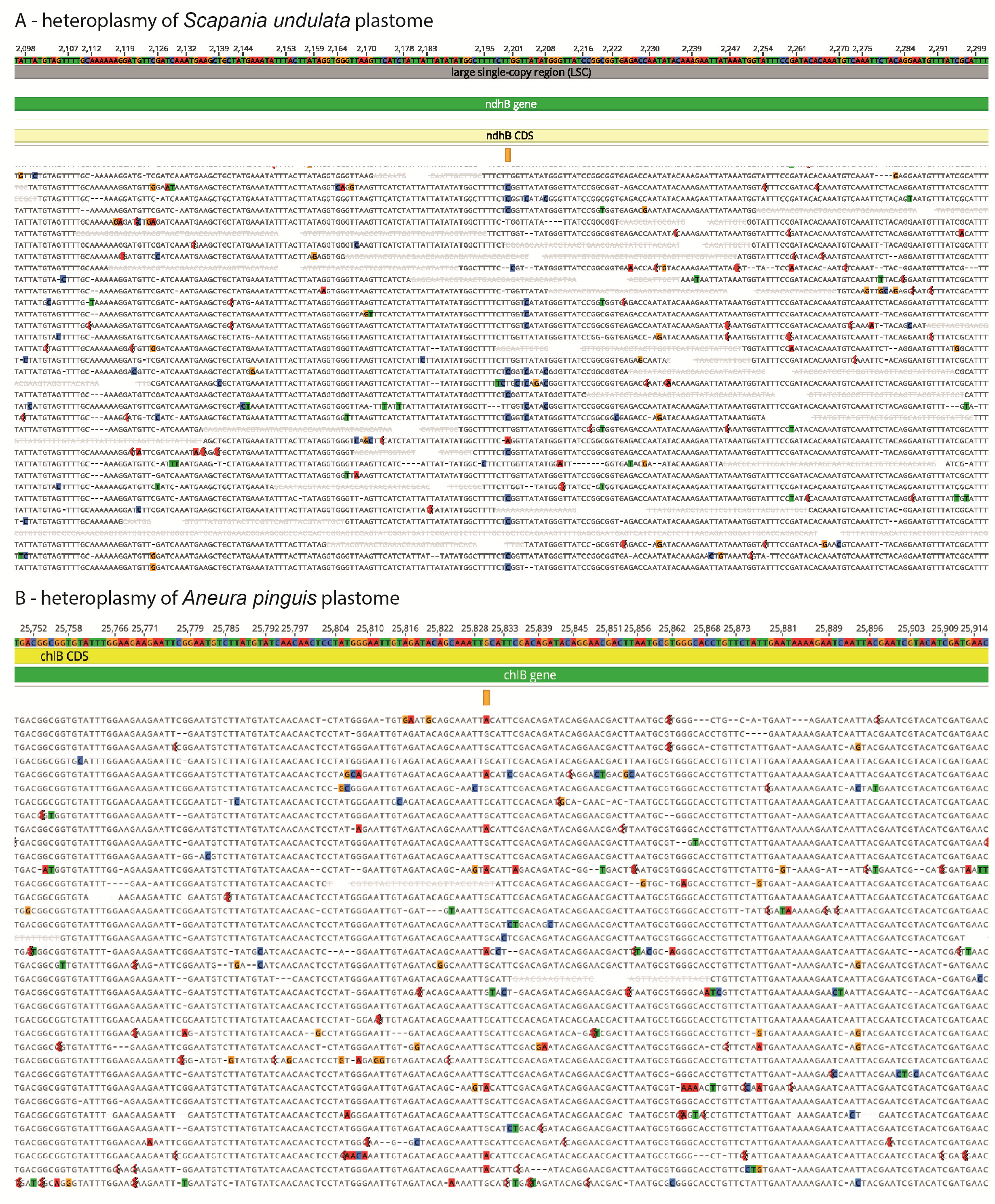
Figure 3
The circular presentation of the heteroplasmic regions within the plastid genomes. The scatter dots indicate substitutions within Scapania (red) and Aneura (blue) plastomes. The shade larger dots show nonsynonymous substitution within CDS regions. The histograms depict the number of deletions (green), insertions (red), and substitutions (blue) identified as a heteroplasmic marker.
. Intraindividual variation of liverwort mitogenomes
Similar to the plastome, the analysis of Riccia fluitans mitogenome did not reveal any instances of heteroplasmy.
Analysis of intraindividual variation of mitogenome within Scapania undulata revealed the presence of 91 polymorphic sites, including nine indels and 82 substitutions. Among detected indels, two were located in the intronic region of cox1 and atp9 genes, while remaining in intergenic regions. All indels can be qualified as short sequences repeat polymorphism (SSR), including two mononucleotide variants (but differing by 5 bp), six dinucleotides (CT and AT motifs) and one trinucleotide variant with AAT motif (Figure 4A). The transitions (65) dominated over transversion (17), but both types of substitutions were mostly found in non-coding regions. In the protein-coding genes, only three transversions and seven transitions were identified within sdh3, rpl2 (two SNPs), cox2 (two SNPs), rtl, nad9, ccmFC (two SNPs) and ccmFN genes. Four of these substitutions found in rpl2, nad9, rtl and ccmFN genes were non-synonymous.
Figure 4
Examples of mitochondrial heteroplasmy in Scapania undulata (A), Apopellia endiviifolia (B) and Aneura pinguis (C).
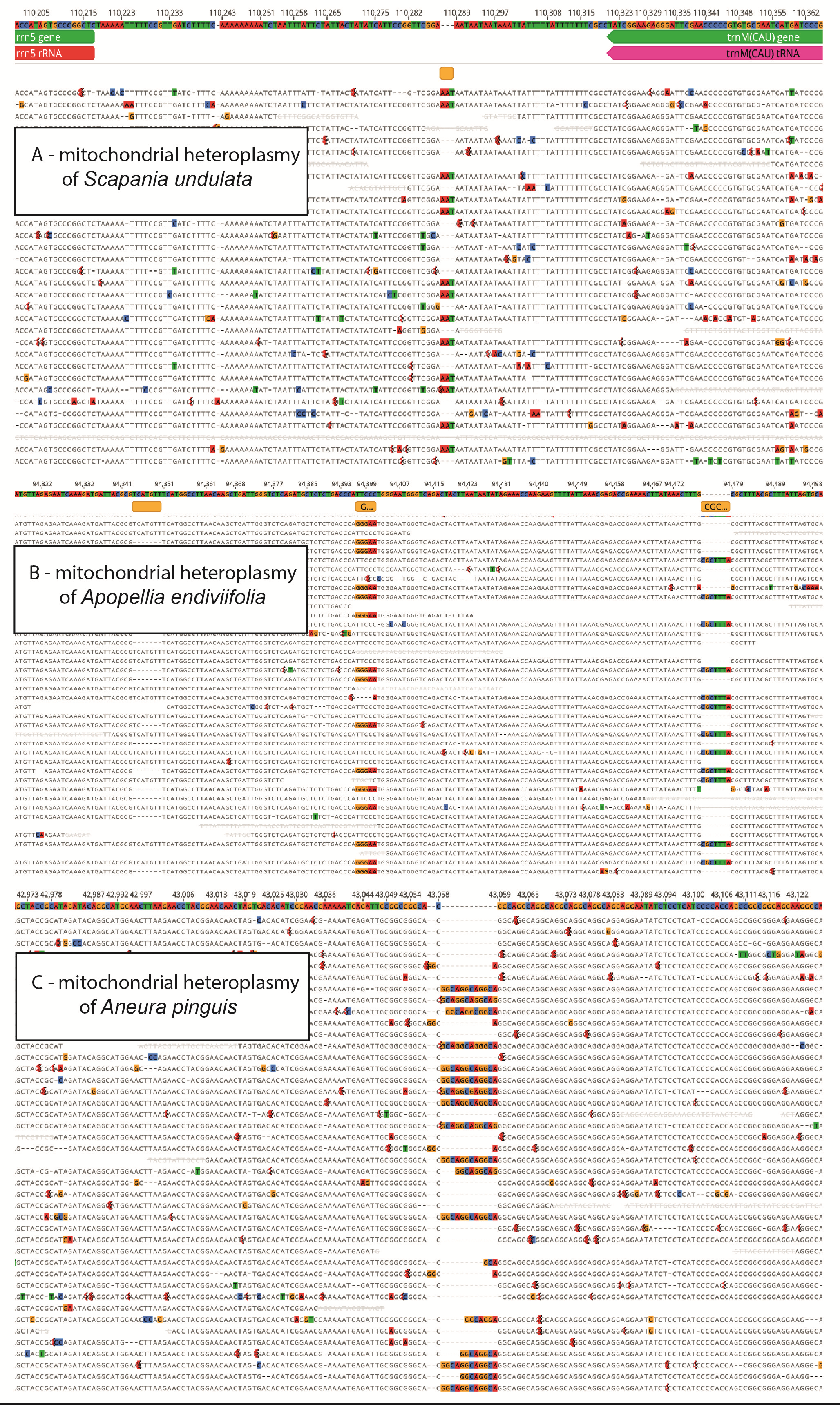
Intraindividual variation of Apopellia endiviifolia mitogenome comprised mainly indels (475), but three substitution events were also detected. All those substitutions were found in non-coding regions and can be qualified as multiple nucleotide variants (MNV), causing changes that concern five to six adjacent nucleotides (Figure 4B). Among indels, 42 were identified within CDS, 30 of them cause potential frame-shift, while the remaining expand or shorten product up to three amino acids. These indels not causing frameshift were found across several genes, including rpl2, rpl5, rps3, rps7, rps19, cob, ccmFC, ccmC and sdh4. Altogether, only 19 protein-coding genes (atp6–9; cox1–3; nad2–5; rps1, 8, 11–14; rpl10, 16) remained unaffected by intraindividual indels. Besides typical SSR polymorphism, including mono-, di-, tri-, and tetranucleotide, the character of identified indels is very specific - most of them are 7–14 nt long sequences that are duplicates or triplicates of adjacent regions.
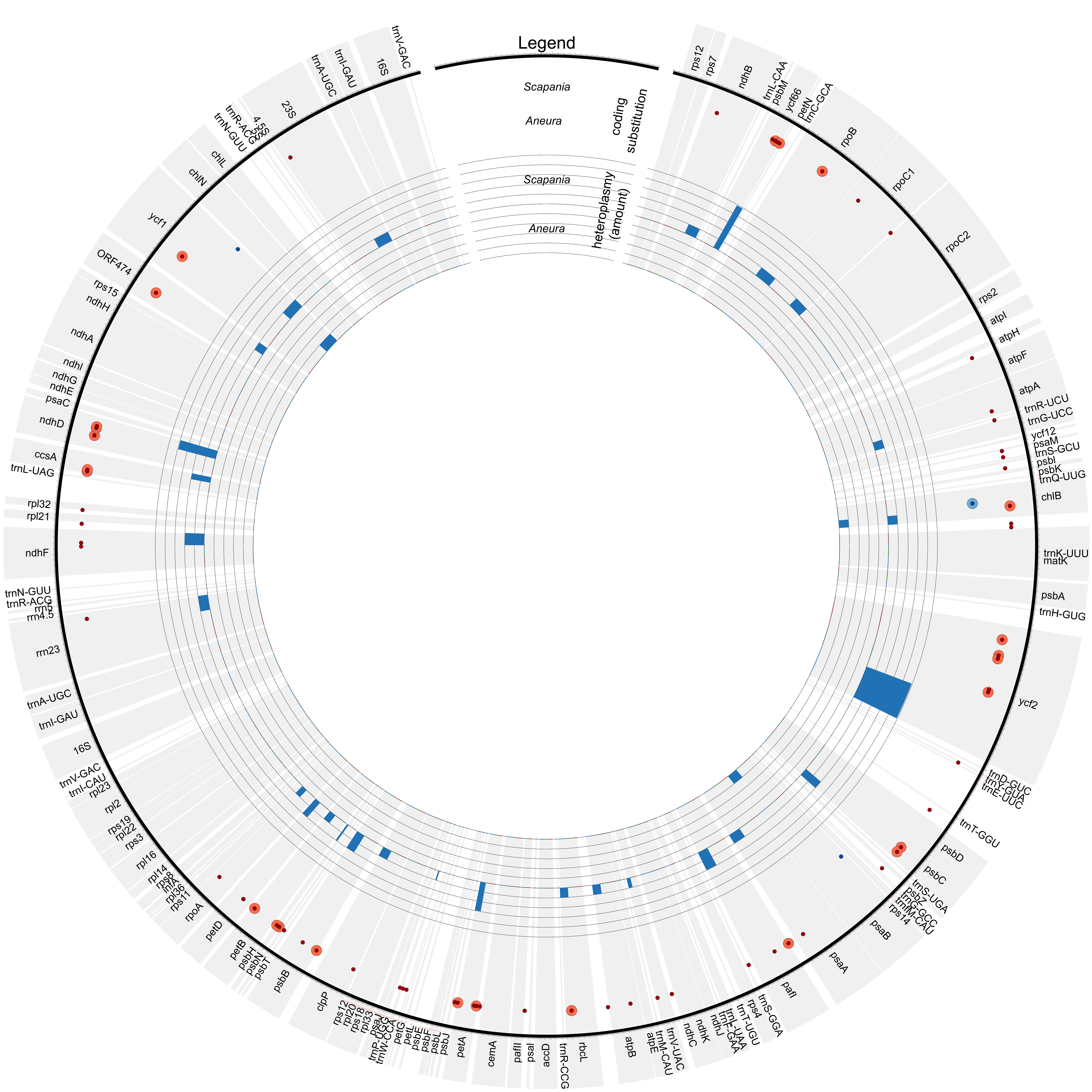
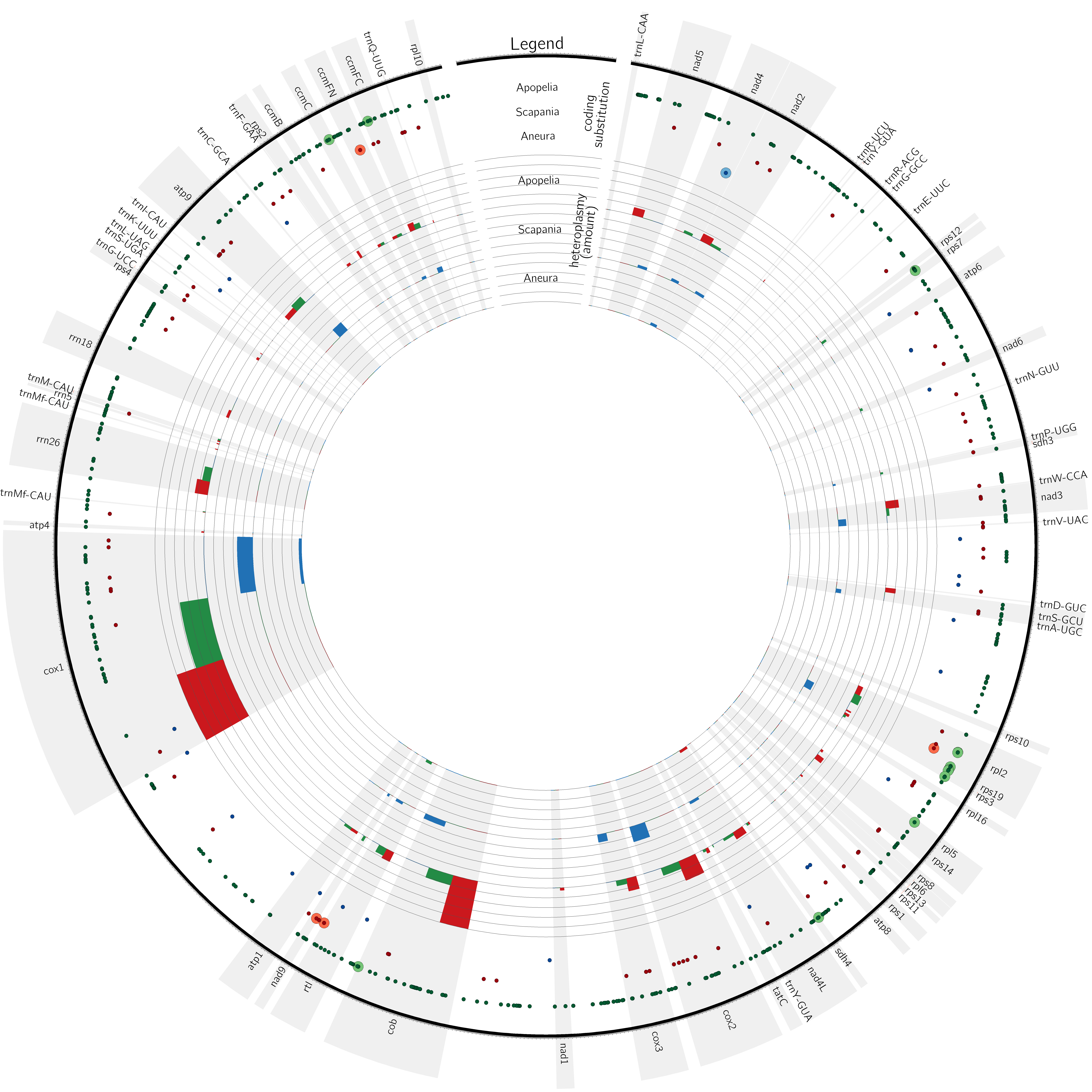
The detected heteroplasmy within the Aneura pinguis mitogenome represented both indels and substitutions. Most of the indels, with the exception of a single “GA”, were of SSR type, including three loci of TA and two CT dinucleotides repeats (TA), and seven trinucleotide repeats of “GGC”, “CGG” and “CGC” motifs. Besides the tetranucleotide SSR motif “CAGG” located in the trnA(UGC)-rps10 intergenic spacer, which was repeated 7–10 times (Figure 4C), the remaining loci differed by one repeat of each motif. The analysis revealed 12 substitutions, including ten transitions (equal proportion of C->T and G->A) and two transversions (both T->A). The latter resulted in one non-synonymous change within the nad4 gene, while the remaining were located within intronic or IGS regions. The distribution of heteroplasmic loci over mitogenome revealed the difference between substitution and indels. While the former were equally distributed, the latter were present mostly in protein-coding regions of the mitogenome (Figure 5).
Figure 5
The circular presentation of the heteroplasmic regions within the mitochondrial genome. The scatter dots indicate substitutions within Apopelia (green), Scapania (red), and Aneura (blue) mitogenomes. The shade larger dots show nonsynonymous substitution within CDS regions. The histograms depict the number of deletions (green), insertions (red), and substitutions (blue) identified as heteroplasmic markers.
. Discussion
. Application of nanopore sequencing in liverwort organellar genomics
The conserved structure and gene content of liverwort organellar genomes are well-known (Dong et al., 2021; Liu et al., 2014; Myszczyński et al., 2017; Ślipiko et al., 2017), and the results of our analysis support this observation. Newly assembled organellar genomes have identical gene order, structure, and content as previously published genomes assembled using short-reads technology. The consensus sequences of organellar genomes of Riccia fluitans, Aneura pinguis, Apopellia endiviifollia, and plastome of Scapania undulata differ by only a few SNPs from previously published (Ciborowski et al., 2022; Min et al., 2020; Myszczyński et al., 2017; Sawicki et al., 2020, 2021) and newly assembled mitogenome of Scapania undulata share the genomic features with S. ampliata (Choi et al., 2021) and S. ornithopodioides (Dong et al., 2019). The mitogenomes of liverworts are characterized by low variation at a generic level, at least considering their structure, gene, and intron content, but this observation is based on limited resources since only a few genera were analyzed (Myszczyński et al., 2017; Ślipiko et al., 2022). A similar situation was also observed in liverwort plastomes (Myszczyński et al., 2017; Ślipiko et al., 2020, 2022) with the exception of Conocephalum, where IR expansion was observed in C. salebrosum (Sawicki et al., 2020).
In contrast to research on angiosperm chloroplast genomes, previous studies on Scapania undulata (Ciborowski et al., 2022), Apopellia endiviifolia and Pellia epiphylla using long-read sequencing (Paukszto et al., 2023) did not detect structural heteroplasmy associated with SSC subunit orientation. Long-read sequencing revealed a single plastome haplotype in gymnosperms and pteridophytes, suggesting that the alternative haplotype is specific to angiosperms (Wang & Lanfear, 2019). However, the number of liverwort organellar genomes assembled using long reads is still too low to exclude the possibility of structural variation. Most of the available plastomes were assembled using Illumina technology (Dong et al., 2021; Myszczyński et al., 2017; Yu et al., 2019), which makes detection of structural variants difficult. Recent advances in nanopore sequencing, including 10.4.1 pores, duplex reads, and improved basecalling enabled fast and high-quality assemblies of plastomes without the need for polishing with short reads. Moreover, long-read technologies have overcome the shortcomings of short-read technology and enabled the assembly of complete organellar genomes in species rich in short tandem repeats, like Conocephalum (Sawicki et al., 2020), Apopellia and Pellia (Paukszto et al., 2023).
. Heteroplasmy of plastid genomes
Numerous studies have been conducted to explore plastome heteroplasmy in vascular plants. Earlier studies applied fragment length variation of selected plastome regions (Ellis et al., 2008; Iida et al., 2007). Additionally, heteroplasmy has also been identified through SNP analysis obtained by next-generation sequencing. This approach demonstrated the presence of heteroplasmy in both chloroplast and mitochondrial genomes of Phoenix dactylifera L. in a range of 18 to 25 SNPs in mitogenomes and one to eight SNPs in plastomes across various cultivars (Sabir et al., 2014). Structural heteroplasmy of the plastome has been discovered in species within angiosperms, gymnosperms, and pteridophytes using long-read sequencing (Wang & Lanfear, 2019). It is interesting that most angiosperms have shown heteroplasmy with almost equal proportions of two different haplotypes (haplotypes A and B), and one plant, Selaginella tamariscina, contained a third haplotype (C). The detected infra-individual haplotypes differed by the orientation of the SSC subunit (Wang & Lanfear, 2019). Also, robust structural heteroplasmy was detected in Eleocharis species and it was not related to IR regions (Lee et al., 2020). So far, there has been no information about plastome structural heteroplasmy in liverworts and other bryophytes, and the use of nanopore long-read sequencing in this study also did not reveal any structural plastome rearrangements within the analyzed species. It seems that structural heteroplasmy caused by a difference in SSC orientation is limited to seed plants (Ciborowski et al., 2022; Wang & Lanfear, 2019).
Although the phenomenon of plastome heteroplasmy in vascular plants has been described by many scientists (Mandel et al., 2016; Sabir et al., 2014), there are no studies about heteroplasmy in bryophytes. The analysis of plastid genomes of four liverwort species representing all main evolutionary liverwort lineages indicates the presence of heteroplasmy in simple thalloid (Aneura pinguis) and leafy liverwort (Scapania undulata). At this stage and with limited available PCR-free libraries, it is hard to conclude if molecular mechanisms behind heteroplasmy evolve after the divergence Pellidae and Metzgeriidae/Jungermanniidae lineages.
. Mitochondrial heteroplasmy in liverworts
Heteroplasmy also occurs in plant mitochondrial genomes, mainly due to recombination among large repeats, which leads to changes in the structural organization of the mitogenome (Gualberto et al., 2014). Arabidopsis thaliana, Nicotiana tabacum, Triticum aestivum have shown recombinogenic repeats in numbers of two, three, and ten, respectively (Klein et al., 1994; Ogihara et al., 2005; Sugiyama et al., 2005). Except for circular genomes, linear, branched, and multi-chromosomal forms were also found in many species (Allen et al., 2007; Kozik et al., 2019; Szandar et al., 2022). In liverwort mitogenomes, despite the presence of some repeats, recombination events are very rare, currently only known from two species, the leafy Gymnomitrion concinnatum and the complex thalloid Dumortiera hirsuta (Kwon et al., 2019; Myszczyński et al., 2018).
Some of the detected substitutions in the organellar genomes of Scapania undulata and Aneura pinguis share a common pattern of RNA editing, which is common and sometimes very intensive in liverworts (Dong et al., 2019; Myszczyński et al., 2017). RNA editing is a process that modifies transcripts from both organellar and nuclear genomes in various organisms such as animals, plants, fungi, and protists (An et al., 2019; Knoop, 2011; Rüdinger et al., 2011; Teichert, 2018). RNA editing is a crucial mechanism that supplements the central dogma by producing RNA products that differ from their DNA templates. The primary form of RNA editing in plants involves the conversion of C-to-U (canonical) or U-to-C (non-canonical) in mitochondria and plastids. This modification generates alternative amino acid sequences and is necessary for the proper functioning of some protein-coding genes (Ichinose & Sugita, 2016). In plants, RNA editing is more common in mitogenomes than plastomes (Ruwe et al., 2013), but the frequency and the type of edition are species-specific (Rüdinger et al., 2012). Transcripts containing edited nucleotides can be transferred to organellar genomes via retroprocessing. According to multiple reports (Cohen et al., 2012; Derr & Strathern, 1993; Fink, 1987), retroprocessing, also known as the reverse transcriptase-mediated model (RT-mediated model), is the most commonly observed mechanism for removing introns. This process involves the reverse transcription of spliced and edited mRNA, followed by the integration of the resulting intronless complementary DNA (cDNA) into the genome by homologous recombination (Cohen et al., 2012; Derr & Strathern, 1993; Fink, 1987). As a result, the loss of introns is accompanied by the loss of editing sites in the mitogenomes and plastomes, and this process is known from liverworts (Dong et al., 2019; Ślipiko et al., 2017).
This mechanism can explain the lack of heteroplasmy in Riccia fluitans, which, like other complex thalloid liverworts in non-editing species (Dong et al., 2019; Myszczyński et al., 2019), as well as some transitions in other species, especially in Scapania undulata (Figure 2). Moreover, the evolutionary rate of complex thalloid organellar genomes is the slowest among main liverworts lineages (Sawicki et al., 2020; Villarreal et al., 2016; Xiang et al., 2022), which could also impact intraindividual variation.
Apart from leafy S. undulata and simple thalloid Aneura pinguis, the heteroplasmy detected in the mitogenome of Apopellia endiviifolia is based on copy number variation of short repeats present in protein-coding and non-coding regions. Some studies suggest that this kind of copy number variation can be an artifact of PCR enrichment and PCR-based sequencing methods (Nakai et al., 2019), however, in our study, we used the native DNA sequencing method, which did not involve PCR steps. Generating short repeats is mainly explained by replication errors due to polymerase slippage and inefficient DNA repair processes (Fan & Chu, 2007). Mitogenome evolution in Pellidae (the subclass of A. endiviifolia) is the fastest among liverworts (Paukszto et al., 2023), which could also be correlated with high mutation rates. In the case of mitogenomic heteroplasmy within Apopellia, the mechanism related to biparental inheritance could be excluded, since the plastome of this taxa lacks any intraindividual variation.
. Conclusions and future perspectives
The nanopore sequencing is able to clearly image the phenomenon of heteroplasmy. Especially in plants, where both plastid and mitochondrial genomes show big numbers of single nucleotide polymorphisms. This study verified that heteroplasmy is also present in liverworts, not only in vascular plants. It also confirmed the conservative character of the liverwort’s plastome because no changes in structures and no structural heteroplasmy were found. However, further studies are required to better understand the heteroplasmy of liverwort organellar genomes. Besides wider taxon sampling, including dioecious, monecious, and vegetative propagative species, transcriptomics and deep analysis of replication surveillance genes could help to explain this phenomenon.
